
Industry insight: applying QbD principles for AAV sterilization filter selection in drug substance clarification and final drug product processing
In the development of gene therapies, the adeno-associated viral vectors (AAVs) have garnered significant interest for numerous applications because of their broad host infectivity range and acceptable safety profiles. Successful clinical translation and market approval of AAV-based therapeutics hinges on developing robust AAV manufacturing processes that can produce products with the desired characteristics critical to product quality and patient safety.
Defining the appropriate Chemistry Manufacturing and Controls (CMC) strategies for AAV-based gene therapies is one of the biggest obstacles facing developers and presents a significant risk to the success of new Investigational New Drug (IND) applications for potential candidates. To avoid setbacks, regulatory bodies like the FDA have been advocating the Quality by Design (QbD) approach to fulfill regulatory expectations for CMC information.
In the article, Consideration of quality by design principles in the use of sterilizing grade filters for clarification and final sterile filtration in adeno-associated virus applications written by Pall Corporation and published by Cell and Gene Therapy Insights, expert authors provide guidance and outline recommendations based on QbD principles for the successful implementation of sterilizing grade filters in AAV manufacturing processes. Sterilizing grade filtration plays an important role in AAV manufacturing, both for drug substance (DS) as well as final drug product (DP) to ensure patient safety. The recommendations and guidance outlined in the article are based on experimental analysis, insider insight derived from projects executed through Pall’s AcceleratorSM Validation Services Program and current CMC requirements as outlined by regulatory agencies.
What is QbD?
The goal of QbD is to build quality into manufacturing from the outset rather than relying on final quality testing alone and should be implemented during early stages of development. The first step is to define a quality target product profile (QTPP) which includes specifications of safety, purity, and efficacy of the drug product. Detailed understanding based on both prior knowledge and experimental data is needed to identify these critical quality attributes (CQAs). Once these objectives have been outlined, the design space for critical process parameters (CPPs) and critical material attributes (CMAs) that impact CQAs can then be established. The aim is to ensure CQAs are maintained within acceptable limits by implementing an effective manufacturing control strategy to keep CPPs and CMAs within their design or control space. With respect to sterilizing grade filtration, the relevant CQAs, CPPs and CMAs are outlined in Table 1.
Table 1. Summary of most relevant CQAs, CPPs, and CMAs for sterilizing grade filtration in AAV DS and DP manufacturing.
| Impacted QbD criterion* | Application | |
|---|---|---|
| Clarification (DS) | Final sterile filtration (DP) | |
| COA | Bioburden Functional AAV titer Aggregated AAV Residual host cell DNA1 Residual host cell protein1 Endotoxin2 Leachables3 |
Sterility Functional AAV titer Aggregated AAV Leachables3 |
| CCP | Differential pressure Flux |
Differential pressure Throughput/flux decay Flux Duration |
| CMA | Filter capacity Conductivity |
Filter performance Filter robustness |
The relative infancy of the gene therapy market means that CMC experience is limited, with only a handful of approved drugs developed for small patient populations. As well, many regulatory agencies have frameworks in place to accommodate expedited clinical development of gene therapy treatments, which can restrict opportunities (due to limitations in time and product availability) to develop CMC information more robustly using QbD principles. Because of these factors, the authors highly recommend early engagement with regulatory agencies and establishing key partnerships with suppliers to increase the probability of regulatory acceptance of the submitted CMC documentation, especially when scaling up the process to keep pace with rapid progress in the field.
Application QbD principles to sterile filtration
Although the use of sterilizing grade filters is well understood for many traditional pharmaceutical applications, AAV-based gene therapies present some unique challenges for manufacturing. Sterilizing grade filters are considered a raw material for gene therapy products because they come into direct fluid contact with the DS / DP. Therefore, during early phase clinical development, it is critical that a filter is selected that meets the quality requirements for a pharmaceutical product. Specifically, for AAV DP, choosing to use a non-pharmaceutical grade filter for Phase 1 clinical batches can impede the incremental development of CMC information especially if it cannot be integrity tested in later clinical stages. There is a risk that non-pharmaceutical grade filters will not meet the quality requirements necessary for processing drug products (i.e., cleanliness requirements or level of manufacturing quality control) and having to switch to a filter that meet specifications after initiating clinical trials may require significant re-work that can delay regulatory approval.
In a typical AAV manufacturing process, sterilizing grade filters are commonly included after depth filtration during AAV harvest clarification. This effectively removes cellular debris and other particulate matter from the DS and reduces the potential bioburden and risk of introducing endotoxin in the process. During the final formulation and filling of the heat labile AAV, sterilizing grade filters are used to assure the sterility of the DP. It is important to note that in addition to detailed CMC information, full sterilizing grade filter validation testing is required to support a Biological License Application (BLA) or Marketing Authorization Application (MAA).
Regardless of which stage it is used, sterile filtration must be controlled to create a robust manufacturing process that assures control of CQAs for the DS and DP. And while the CQAs for DS and DP are different, the key factors to consider for selection of a sterilizing grade filter are similar. The authors provide a list of key criteria for consideration during the filter selection step (Table 2), a compilation that is based on Pall’s experience and/or known regulatory requirements, and best practices for the manufacturing process.
Table 2. Factors to consider for selection of a 0.2 µm sterilizing grade filter for an AAV manufacturing process and potential criticality (high, medium, low) as applied to clarification (DS) or final sterile filtration (DP). (1)
| Attribute |
|
||||||
|---|---|---|---|---|---|---|---|
| Filter physical properties | |||||||
| Materials of construction |
|
||||||
| Filter effective filtration area (EFA) |
|
||||||
| Retention rating |
|
||||||
| Membrane architecture |
|
||||||
| Number of membrane layers |
|
||||||
| Mode of sterilization |
|
||||||
| Scalable product portfolio |
|
||||||
|
Filter variability
|
|
||||||
| Filter compliance claims/CMAs | |||||||
| Cleanliness (particulate release) |
|
||||||
| Biological reactivity testing |
|
||||||
| Compliance with appropriate regulatory standards (e.g. animal spongiform encephalopathies) |
|
||||||
| Traceability |
|
||||||
| Extractables/leachables |
|
||||||
| Supplier quality dossier |
|
||||||
| Filter performance claims | |||||||
| Performance correlated to integrity test values |
|
||||||
| Bacterial retention claims |
|
||||||
| Operating parameter ranges |
|
||||||
While it is the supplier’s responsibility to ensure that the appropriate sterilizing grade meets the requirements for pharmaceutical use, the filter selection will be based on risk assessments and specific processing needs identified by the end user. Selection of the final sterilizing grade filter for DP should be documented in a risk assessment, with a description of the appropriateness of the equipment for the intended purpose. As the authors stress, not only is this a regulatory expectation, but the risk assessment is also expected to be a living document that is regularly updated as further process knowledge regarding the filtration step is gained.
Sterilizing Grade Filters in AAV Harvest Clarification (DS)
In applying QbD for AAV clarification step, the differential pressure and flux are considered the filtration CPPs, and filter capacity (throughput) and conductivity could be considered CMA to ensure the AAV purity and potency CQAs.
Working within this design space, suppliers recommend suitable filter candidates in different filter configurations that are assessed in bench-scale process development studies with representative clarified harvest (or representative clarified harvest lysate, if lysis is used as part of the process) to evaluate the impact of potential CMAs and CPPs on the CQAs. Throughputs, differential pressures, titers, and any other results can then be compared to select the optimal filter type (membrane material, size), number of filters and configuration based on performance analysis.
These evaluations are critical especially if the end user plans to implement the sterilizing grade filter as part of single-use technology (SUT), a growing trend in bioprocessing to alleviate the need for cleaning validation, increase flexibility and sterility assurance. That’s because SUT assemblies are usually designed with a maximum operating pressure, which can impact the filter selection (e.g., operational or differential pressure limitations for the filters). Consequently, it is vital to review the bench-scale performance data and ensure the differential pressure across the entire clarification filter system (depth filters and sterilizing grade filters in-line) does not exceed the maximum operating pressure of the SUT, which then may become a CMA.
Additionally, peristaltic pumps, which are commonly used for the clarification step, can reach an operational pressure where the pump rollers may start slipping and cannot deliver a constant flux. Therefore, the differential pressure as a CPP across the whole clarification filter system must be less than the maximum operational pressure for the SUT and below the operational pressure that the pump rollers may start slipping. Consideration should be given to implementing an adequate safety factor (Proven Acceptable Range, PAR), which the end user should base on risk assessment and the level of manufacturing control associated with the normal operating range (NOR).
The authors highlight other variables in the AAV process that can also impact filtration during clarification, either individually or in a multi-combination effect that include (but are not limited to) choice of cell line, suspension vs. adherent culture, viability, AAV serotype, titer and more.
While the body of knowledge regarding suitable depth and sterilizing grade filter combinations for AAV clarification is growing, a greater comprehension of the impact of these variables on the filter performance (CPPs and CMAs) is needed before a platform technology approach could be considered that would allow for consistent performance and acceptable AAV titers during clarification. The authors describe experimental data to better describe the clarification design space during the sterilizing grade filter step (post-clarification).
Feed material was generated in a five-day production process in three separate bioreactors. Cells were lysed to release intracellular AAV vector and treated with DNA endonuclease to remove free DNA contaminants. Each bioreactor harvest lysate was clarified with the same depth filter combination to generate clarified harvest lysate for subsequent sterilizing grade filter testing: Supor® EKV sterilizing grade membrane or Supor EX ECV sterilizing grade membrane. Data is presented in the article showing the normalized AAV step titer (based on ddPCR) for clarified harvest lysate and the minimum/maximum throughput and differential pressure values from a range of the bench-scale experiments.
Sterilizing Grade Filters in Final Sterile Filtration (DP)
Final drug product sterility is an important CQA for patient safety; therefore, sterilizing filtration is a requisite in the DP manufacturing process. The final filter should be classified by the filter supplier as “sterilizing grade” and be rated at 0.2 µm or less. This classification confirms that the filter meets the regulatory requirements of being 100% retentive for a standard bacterium (Brevundimonas diminuta ATCC 19146) under defined conditions (as per ASTM F838-20) at a challenge level of ≥ 1.0 × 107 CFU/cm2.
Filterability testing is typically performed during initial filter selection with different membrane types and under several differential pressures and/or flow rates on scaled down devices to determine suitable filter sizing for expected throughput. AAV aggregation, filter adsorption and overall AAV titer should also be evaluated post-filtration during the tests. While in depth knowledge of potential CPPs with respect to final sterile filtration is unlikely during early phase clinical trials, it is critical to show regulators that even with gaps in the CMC knowledge, the therapeutic DP produced will not present safety concerns to the patients. Early engagement with filter suppliers can be advantageous since their expertise can help with filter selection based on regulatory and process requirements.
As process development continues, knowledge regarding the potential design space for final sterilizing grade filtration for AAV DP should increase as well. Ideally, this is achieved through design of experiment (DoE) to understand the interaction of CPPs and CMAs, with throughput, flux and/or differential pressure being the most important CPPs to consider for their potential impact on AAV CQAs such as aggregation and titer.
To evaluate a potential sterile final filtration design space for AAV applications, a review of thirty-two AAV gene therapy projects from Pall’s AcceleratorSM Validation Services program was performed to identify industry trends for CPPs used to define PARs. The first observation noted by the authors is that many end users requested filter validation studies very early on in process development, even though they are not required by regulators for first in-human clinical trials (Figure 1).
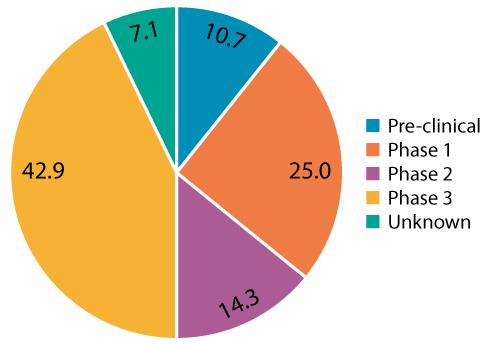
The authors caution that executing these studies too early can pose a risk since potential changes may occur during further scale-up or technology transfer. For early process development, confirmation of sterilizing grade filter performance by post-use integrity testing is typically sufficient. When polled, some of the reasons for these early requests include:
- The end-user was transferring the process to a contract manufacturing organization (CMO), and was advised filter validation was required to support the transfer
- The application was designated an orphan drug and the manufacturing process for low volume batch sizes was not expected to change
- The application was on a fast-track regulatory pathway
Ultimately, the decision on when to perform final sterilizing grade filter validation studies is based on the risk assessment, but other factors including product availability for the testing, and the likelihood of a change to the manufacturing conditions as process development continues, should be factored in.
Other notable takeaways described by the authors in reviewing the final filter validation projects for DP include:
- 90% of end users selected a sterilizing grade filter composed of Polyethersulfone (PES) membrane
- Final sterilizing grade filters had small effective filter areas (EFAs) ranging from 20 to 220 cm2
- Approximately two thirds of end users used a constant (and low) flux to control the final sterile filtration step to minimize shear, reduce the potential for AAV aggregation and loss in functionality
- A broad range of filtration times was observed with most end users adding a significant safety factor to accommodate the time for the final post-use integrity test or to account for disruptions during the final sterile filtration step
The data analysis revealed that for AAV DP, final sterile filtration is performed using CPPs at a lower range—lower flux, shorter filtration times, smaller batch sizes—compared with the validated design space for other established biological DP. The range of CPP tested and validated in the studies does support a robust design space, leading the authors to conclude that final sterile filtration of AAV DP is a low-risk unit operations from a sterility assurance perspective.
A keen understanding of the interaction of the CPPs and CMAs on the drug product CQAs, as well as having an appropriate control strategy to maintain parameters within their acceptable limits, is required to fully define the sterile filtration design space according to QbD principles. The authors hope that the experimental and retrospective data analysis information shared in the article will provide guidance for future AAV-based therapies and enable the development of platform technologies that can be implemented in the long term as the as the field continues to mature.
To download a copy of the article, please visit: Consideration of quality by design principles in the use of sterilizing grade filters for clarification and final sterile filtration in adeno-associated virus applications