
The need of a therapeutic gene editor to develop the next generation of CAR T Therapy
Two chimeric antigen receptor (CAR) T-cell therapies (Kymriah® and Yescarta®) are approved by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) to treat B-cell acute lymphoblastic leukemia in children and young adults and diffuse large B cell lymphoma in adults, respectively. Both therapies resulted in high objective response rates (~80% of treated patients) during clinical trials (Neelapu, 2017) (O’Leary, 2018). Despite these approvals, questions remain over whether cell therapies, such as CAR-T-cells, represent a new era of frontline therapy with potential significance to match that of monoclonal antibodies or whether they will be an effective but niche approach for a narrow range of indications and a relatively small number of patients. This is primarily owing to the challenges encountered when treating patients with solid as opposed to liquid tumors. It is likely that CAR T-cells will need to up their game through genetic engineering if they are to have efficacy in the notoriously immunosuppressive solid tumor microenvironment.
From approved CARs to universal CARs
Currently, CAR-T therapies are constructed using the patient’s own T cells, where on removal from the patient, the T cells are expanded and transduced with a CAR, which binds a specific protein on the surface of the tumor cells. Therefore, when infused back into the patient, the CAR can recruit the T-cell to a tumor cell and allow it to engage with that cell. Engagement sends a signal to the CAR T-cell to attack the attached cell, leading to the death of the tumor cell. In this paradigm, the CAR acts almost as a targeting system for the effector component of the therapy, which is the T cell.
The process of generating the CAR T cells described above and used to generate both Kymriah and Yescarta is known as autologous cell therapy. This process takes time, has demonstrated issues with process control and is one of the major contributors to the high treatment price of these therapies. The ‘vein-to-vein’ time to treat patients with these therapies is 2-3 weeks, consisting of several different steps leading to a complex manufacturing and supply chain. In fact, by the end of 2019, Novartis had been unable to ship a product 10% of the time due to manufacturing failures or out of specification issues. As a result, a number of companies are attempting to develop allogeneic or universal therapies, whereby a single source of donor cells is used to create a bank of CAR T cells to treat multiple patients in a process more analogous to traditional biologics manufacturing. However, to develop allogeneic therapies, considerable changes to the T cells need to be engineered.
To date, substantial improvements to the efficacy of CAR-T cells have come from altering the design of the CAR, such that activation of the T cell is improved. However, aside from the expression of the CAR, current approved CAR T-cells are largely unmodified, with the T cell essentially being in its natural state. Modifying the T cell, for example, could improve its capability to target cancer cells by reducing its capacity for exhaustion. Additionally, to enable targeting of solid tumors and therefore move beyond a relatively small patient population served by the approved indications for Kymriah and Yescarta, a number of natural obstacles need to be overcome. For example, numerous cytokines secreted by tumours inhibit trafficking of T-cells and the immune suppressive nature of the tumor microenvironment means that T-cells are less able to exert their cytotoxic effect on tumor cells should they reach the tumour periphery. For allogeneic CAR T cells, additional challenges exist in that these cells could be viewed as foreign by the patient’s immune system and killed soon after delivery. Allogeneic CAR-T cells also pose a risk of triggering graft vs host disease (GvHD), where the engrafted T cells see the patients tissues as foreign and attack them as well as the tumor cells. Thus, allogeneic CAR T cells must be modified so that they persist for long enough to be effective and do not induce GvHD.
What is clear is that there are several different ways in which CAR T-cell therapies can be improved through altering the underlying biology of the T-cell, allowing them to respond differently to intra- or extra-cellular stimuli. As such, responses are often mediated through differential regulation of target genes, methods for modifying specific genes to achieve a desired phenotype is a key area of research within the cell therapy field.
Engineering the next generation of CARs
It is only relatively recently that genome engineering technologies have progressed to the point that it is conceivable to use these techniques in a therapeutic setting. Well known approaches such as, Zinc Finger Nucleases (ZFNs), Transcription Activator Like Effector Nucleases (TALENs) and Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR)–Cas, use similar mechanisms of action to engineer the genome. The most common type of engineering event is knockout of a gene, primarily because from a technical perspective this is the simplest and most efficient way to modify a genome. To modify a gene so that it can no longer produce mRNA and from that protein, traditional editing techniques target a specific DNA sequence and use a nuclease to cleave the DNA and generate a double strand break (DSB). In mammalian cells, these breaks are predominantly repaired using non-homologous end joining (NHEJ), an inherently error-prone mechanism of DNA repair. Often bases will be added or excised from the break point, leading to the creation of insertions or deletions (‘indels’) when the broken ends are re-joined. If these indels change the reading frame of a gene (frameshift mutation), this will often lead to a nonsense mRNA and thereby a functional knockout of the target gene. If targeted correctly, ZFNs, TALENs and CRISPR–Cas editing technologies have high editing efficiencies. Such approaches are being used to improve CAR-T therapies, so why are there not more genetically engineered CAR-T therapies available?
One reason for this is time. Although generation of CAR T-cells is not simple, the first CAR-T therapy approved was always likely to be technically the simplest. As described above, even with relatively few interventions, the manufacturing of this therapy is far from simple. As such, an understandable strategy is to improve on these existing therapies, for example through engineering an allogeneic platform instead of an autologous one using the same effective CAR design present in Kymriah and Yescarta. Indeed, several such allogeneic CAR T cells have entered clinical trials with genome engineering steps included. However, none of these CAR T cells have more than 3 genes modified and some estimate that up to 10 genetic changes could be required to produce a robust, efficacious, and safe allogeneic CAR T cells to treat solid tumors. So why have CAR T cells with higher numbers of altered genes not entered clinical trials?
to be technically the simplest. As described above, even with relatively few interventions, the manufacturing of this therapy is far from simple. As such, an understandable strategy is to improve on these existing therapies, for example through engineering an allogeneic platform instead of an autologous one using the same effective CAR design present in Kymriah and Yescarta. Indeed, several such allogeneic CAR T cells have entered clinical trials with genome engineering steps included. However, none of these CAR T cells have more than 3 genes modified and some estimate that up to 10 genetic changes could be required to produce a robust, efficacious, and safe allogeneic CAR T cells to treat solid tumors. So why have CAR T cells with higher numbers of altered genes not entered clinical trials?
The main reason for this is that of the concern around patient safety because of genome engineering. These safety concerns can be broken down into two main categories. The first is off-target effects. ZFNs, TALENs and CRISPR–Cas engineering platforms have to bind to a specific sequence in the DNA to modify a gene of interest. But there are often other sequences in the genome that are highly similar to the target sequence and are bound by the nuclease in error. In this scenario, additional genes could be knocked out and these could have deleterious consequences for the patient. One obvious example is if the gene knocked out is a tumor suppressor gene — this could lead to the patient being infused with a potentially cancerous T-cell and if that T cell survives long term in a successfully treated patient, that patient could go onto develop a T leukemia driven by the engineered T cell. The risks of off-target effects are currently mitigated by exhaustive analysis of potential off-target knockout generation using a combination of in-silico and lab-based techniques. If any off-target sites are found, then either the engineering reagents need to be re-designed, or a risk assessment performed to analyze the consequences of knocking out the identified off-target genes. This could be done by generating cell models containing deliberate knockouts in those genes to analyze the phenotype generated. However, where multiple off-target loci are identified, the consequence of knocking out different combinations of the genes needs to be assessed as this can be very different to the impact of knocking out genes in isolation. Importantly, there is currently no agreed industry standard for identifying off-target effects and there remain difficulties in establishing comprehensively where any off-target binding occurs.
The second safety concern revolves around the generation of DNA DSBs that are inherent to the efficiency of the technologies. If the DNA is broken in more than one place through introducing DSB in several genes at the same time, the NHEJ repair pathway could re-join the loose ends incorrectly, leading to chromosomal translocations. Although the likelihood is that such aberrations in the genome will lead to the death of the cell, there is a risk that one of these translocations could confer a proliferation or survival advantage to the T-cell, potentially leading to a pro-cancerous cell. The impact translocation is much harder to predict than that of a simple gene knockout, as it is impacted by the precise indel on both sides of the break and whether this generates a functional hybrid protein with cancer causing effects. Such alterations are difficult to model using in vitro techniques, as replicating a precise translocation is technically challenging. The unpredictability of these translocations means that even targeting only two genes could lead to unintended consequences, even though there are only four possible translocations (assuming no off-target events) to attempt to model. If one assumes that multiple genes will need to be targeted simultaneously to substantially improve the efficacy of allogeneic CAR T cells, the prediction of possible translocations becomes exponentially harder. For example, if one were to target only 3 genes and assume 2 potential off-target sites for each engineering event, this would require mapping 768 different translocations to be modeled and this is without allowing for the impact of different frameshifts generated after each indel formation! (See figure 1).
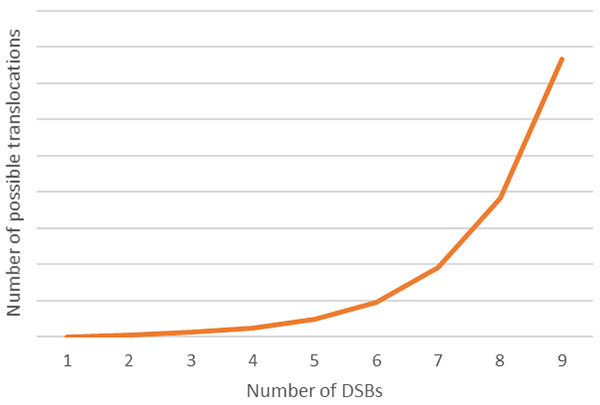
It is important to note that different technologies have different rates of off-target binding and as the significance of off-target editing induced by traditional gene engineering technologies is of concern to developers of cell-based therapeutics, the industry is always looking to improve these technologies. It is not uncommon to hear discussions around 6-10 knockouts being required to achieve the complexity of engineering needed to improve the efficacy of CAR T-cells such that they can impact solid tumors more effectively. If this is done in a single step, assessing the off-target effects effectively rapidly becomes unfeasible, so the question then becomes how to generate this level of engineering without the associated off-target risks.
Addressing the challenge of multiple genetic edits
One of the simplest ways of achieving this is to perform multiple rounds of engineering, thus minimizing the number of breaks occurring simultaneously. However, this has obvious implications around the time taken to generate the engineered cell, and is likely incompatible with an autologous therapy, where a patient with a rapidly advancing cancer does not have weeks to wait for an effective therapy. Allogeneic therapies, particularly those using clonally derived cell lines such as induced pluripotent stem cells (iPSCs) might be more amenable to this approach, but development time is still crucial. Equally, even with only a few engineering steps, the burden of off-target events and consequences on potential translocations is not to be dismissed. Perhaps a more satisfactory solution would be to use a technology that mitigates the risk to patients through minimizing the generation of DSBs and the impact of off-target gene editing.
One such technology that has received significant attention recently is base editing. This technology works by using a deaminase enzyme to carry out a transition mutation that converts one base to another specific base and does not rely upon the introduction of a DNA double strand break. There are naturally occurring cytosine deaminases that convert cytosine to thymine, and recently a synthetic adenosine deaminase has also been developed that converts an adenosine to a guanine. One of the initial therapeutic focuses of base editing was as a tool for gene therapy, with transition mutations believed to be able to address 60% of the disease-causing point mutations that make up many human pathogenic mutations. However, in the context of modifying T-cells to become effective CAR T cells, what is equally important is the ability to use base editing to generate a stop codon, preventing gene transcription and thereby knocking out the gene.
 Current base editing systems work by using a guide RNA and a modified Cas protein that nicks the DNA rather than producing a DSB. The guide RNA brings the Cas to a particular genomic location and through various approaches, this platform can also bring with it a deaminase, either through an interaction of the deaminase with the Cas protein or with the guide RNA. The deaminase, along with specific parts of the host cell DNA repair mechanisms activated by the Cas protein nicking the DNA, converts the target base, in this case a C of CGA codon to a T to generate TGA, a stop codon.
Current base editing systems work by using a guide RNA and a modified Cas protein that nicks the DNA rather than producing a DSB. The guide RNA brings the Cas to a particular genomic location and through various approaches, this platform can also bring with it a deaminase, either through an interaction of the deaminase with the Cas protein or with the guide RNA. The deaminase, along with specific parts of the host cell DNA repair mechanisms activated by the Cas protein nicking the DNA, converts the target base, in this case a C of CGA codon to a T to generate TGA, a stop codon.
What is of course key to this technology is that DSBs are not introduced as part of the gene modification mechanism, so this technologically should have a substantially reduced off-target effect that arise because of introducing DNA DSBs. The lack of DSBs mean that the levels of indel generation are very low so the risk of translocations are proportionately decreased. However, base editing is not devoid of off-target effects and guide-RNA mediated off-target editing can lead to transition mutations in the DNA at loci in addition to the targeted gene. An additional safety factor to consider with base editing is the possibility of guide-RNA independent off-target deamination, where the deaminase targets bases throughout the genome. This possibility is currently being investigated and modifications to the deaminase to limit this capability are being sort.
The approval of both Yescarta and Kymriah to treat patients with B-cell tumors has been a huge event in the history of attempting to use the immune system to treat cancer. Several companies are looking to broaden the immune cells used in a CAR approach and include, but are not limited to, CAR NK-cells and CAR B-cells. These cells have different capabilities compared with CAR T cells, but all require some degree of gene editing to be used effectively in the clinic. Which form of gene engineering will become the front runner for immune cell-based therapies in the coming years is currently open to debate. However, as discussed in this article, base editing could be a route forward to achieving multiple genetic alterations while avoiding potentially deleterious structural alterations to the genome. As more laboratories use base editors to modify cell phenotypes, we will be in a better position to compare datasets and look critically at the strengths and weaknesses of standard gene editing platforms and base editing platforms.
To learn more please visit: https://horizondiscovery.com/en/gene-editing/base-editing
Footnotes
-
1. Neelapu, S. S. et al., (2017). Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. New England Journal of Medicine, 377(26), 2531-2544.
-
2. O'Leary, M. et al., (2018). FDA Approval Summary: Tisagenlecleucel for Treatment of Patients With Relapsed or Refractory B-cell Precursor Acute Lymphoblastic Leukemia. Clinical Cancer Resrach, 25(4), 1142-1146.