
Successful Techniques for Progressing Biotherapeutic Candidates from Discovery to the Clinic
The reality is that many initially promising biotherapeutic candidates never make it to commercialization. It is estimated that only 1 in 1000 preclinical candidates reach the commercial market. Why does this happen? There are many that fail due to lack of efficacy or safety, but there are also candidates that fail due to stability, aggregation, and other issues related to cell line or process issues. While some issues may never be satisfactorily resolved to the point where commercialization is possible, others can be saved with early identification, engineering and process development approaches. Even for candidates that can’t be rescued, it is critical to know this early, so that time and resources aren’t wasted on a product that ultimately has no path forward. Therefore, it is imperative that strategies and technologies are available to evaluate and identify issues early in the development phase and address challenges where possible.
In a recent virtual symposium, Lonza gathered industry experts to provide insight on the resources available for successfully progressing biotherapeutic candidates from discovery to the clinic. Presenters from academia, local biotech companies, and Lonza shared information on new and established technologies enabling translational research. I have highlighted a couple of the talks from the seminar in this article. For more information, Lonza and the MassBioHub are hosting a similar event, “Approaches and tools for maximizing success on the journey from discovery to the clinic,” on October 27th.
Early developability assessment tools – Successful de-risking strategies to support progression of biologics from discovery to development
In this talk, Dr. Yvette Stallwood, Head of Applied Protein Services, Lonza discusses the need for candidates to undergo a developability assessment early in the development process. She stresses the importance of knowing as much information about the product as early as possible. A developability assessment can address many potential issues early; thereby reducing risk and maximizing the chance a candidate reaches market.
Manufacturability Assessment
Dr. Stallwood also discusses the different types of assessments that can be conducted at Lonza as part of evaluating a candidate’s developability. For instance, a manufacturability assessment looks at issues including aggregation and post-translational modifications to assess a product’s ability to be successfully developed and manufactured. The assessment is performed in silico with relatively high throughput allowing up to five candidates to be screened in just 1-2 weeks. These areas are important to look at because post translation modifications and aggregation can impact a product’s stability, efficacy and safety. Lastly, process fitness looks at whether risk can be mitigated via protein engineering, additional control samples, monitoring or can simply serve as a heads up that this may be an issue in the future and the process may need to be modified to accommodate the issue. For example, protein engineering is a tool that can be used to fix issues related to post translational modifications and aggregation. When employed early, protein engineering can significantly reduce the risk of a molecule not progressing to the clinic. She shared that the problem of aggregation is often moved forward to the next group to be addressed. Cell culture teams optimize the upstream process as well as potentially try to increase titer, downstream teams try methods for purification and formulation teams try to formulate around it, however it is a more successful strategy to eliminate it at the source.
Immunogenicity Assessment
An immunogenicity assessment can also be part of a developability assessment. Immunogenicity is the ability of a product to illicit an immune response. It is unwanted in a therapeutic protein where the intention is to exert a specific therapeutic function. Immunogenicity issues can reduce efficacy, compromise the PK/PD and safety, and can lead to severe side effects in some cases. Thus it is an issue that needs to be assessed early. In this way the driver of the immunogenicity issue can be identified. In fact, the FDA has stated that the sponsor should provide an immunogenicity risk assessment as well as a rationale for the immunogenicity testing paradigm in the original investigational new drug application (IND).
Dr. Stallwood walked through the in silico tools available for an immunogenicity assessment. This is often performed in parallel with the manufacturability assessment. At Lonza, using Epibase® in silico prediction for their clients, they look to identify potential T cell epitopes in a protein sequence and rank the highest risk epitopes to provide an overall risk score of the molecule. This ranking can then be compared to other leads or even referenced to marketed products to provide more information about the immunogenicity risk. Dr. Stallwood said there are no “no risk” products, but there are lower risk products and there are tools to possibly reduce the risk depending on the issue. In any case, it allows work to stop early on molecules that are too high risk, thereby saving time and money.
Next, Dr. Stallwood discussed Lonza’s Epibase® in vitro immunogenicity assessment, which uses a human PBMC bank to evaluate the immune response to all aspects of the product. The Epibase® immunogenicity assays assess the human immune response in vitro. Assays are used to assess three main aspects of the immune response – immunotoxicity, innate immune response and adaptive immune response.
Protein Production
Lastly, Dr. Stallwood discussed options for protein production. She explained that Lonza utilizes its LightPath™ Discovery platform to provide material for lead selection and optimization and in vitro expression and characterization. They have a variety of expression platforms including CHO, E. Coli, and Yeast (Pichia).
Protein engineering can also be used for humanization and deimmunization with validation carried out in vitro.
She stated that it is important to keep the mammalian expression platforms the same during early development as what will be used in cell line construction and clinical manufacture, including using the same cell line and vectors. Keeping this consistent reduces the timeline in that time isn’t spent later in transitioning to a different expression system or cell line. Another reason to keep it consistent is that post translational modifications can also vary with the expression system. The Lonza system is completely animal free. They use shake flasks to provide scalability and flexibility.
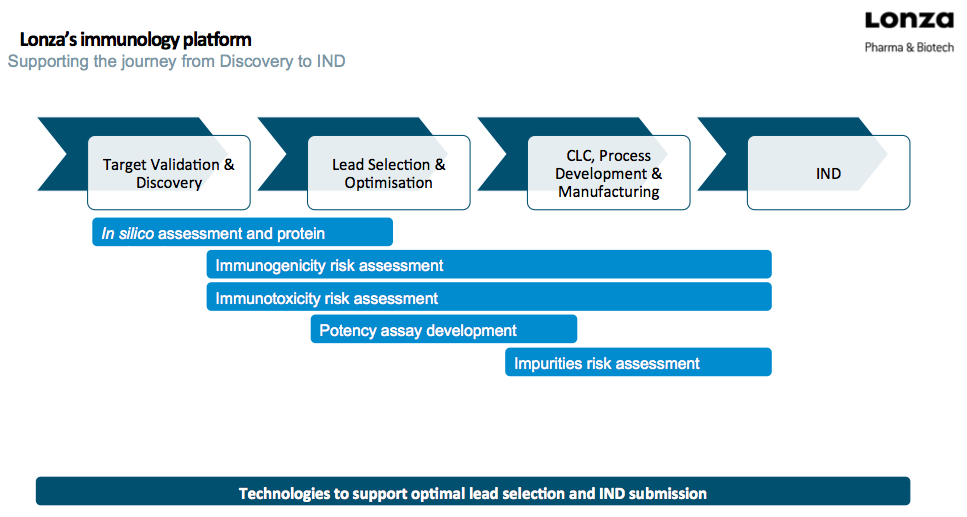
Dr. Stallwood provided a nice overview diagram of Lonza’s immunology platform in Figure 1.
Approaches to maximize successful development of cell lines expressing a diverse range of proteins
Another area that was addressed during the symposium was approaches for maximizing cell line construction. In this talk, Dr. Alison Porter, Head of Expression System Sciences, Lonza discussed how biotherapeutics can fail for reasons unrelated to their mechanism of action or efficacy, sometimes they fail due to problems with the cell line. She pointed out that if the quality of the cell line can be improved, then the hit rate is improved as well.
Dr. Porter addressed the need to move quickly from development to the clinic, but she said that it is important to balance risk with requirements. Going faster when you can, but slowing down when you really need to look deeper at an issue. Not doing so causes real challenges down the road. For instance, with a standard mAb, you can move faster in the process, but the more complex the protein is, the more it is worth taking extra time upfront so as to not encounter problems later.
There are definitely risks associated with accelerating the timeline in early development. In fact, she shared that 47% of Complete Response letters from the FDA name deficiencies in CMC requirements. Risks include a lack of cell line robustness, variable process performance due to poor process design, difficulty scaling up the process to commercial scale, and inadequate or inappropriate drug product design. This can have serious consequences for the success of a product including delays in regulatory approval; need to rework the program stages resulting in increased program cost and length, and difficulties moving quickly to later stages.
Dr. Porter then shared how Lonza helps clients with cell culture platform approaches to successfully deliver cell line construction, process transfer and optimization, and process characterization programs. Specifically with cell line construction, there are four platform options discussed. The first option is a full scope process that provides a thorough, customizable approach to find the special clone. It includes three rounds of screening and assesses a high number of cell lines. Another option is Light Path™, also customizable, but focuses on finding something that works quickly with two rounds of screening and medium level of cell lines assessed. Their Ibex® Design platform is unique in that it guarantees time and quantity. It is only for standard mAbs and mAb-like molecules, but it provides a guaranteed timeline – 12 months to drug product material supply. Lastly they have a Multiplex platform that is customizable and is best for dealing with uncertainty. It offers two rounds of screening and a low number of cell lines screened.
She then detailed Lonza’s GS Gene Expression System, which is a robust, fully integrated system with a proven track record of successfully taking programs from gene through cell line construction to commercial product. It includes tools such as CHOK1SV GS-KO® and other host cell line options, multiple vectors, GS piggybac™ transposon technology, as well as a GS media and feed platform.
Dr. Porter walked through some examples of cell line construction workflows used at Lonza and provided instances where time might be saved, but risk was increased. She also discussed areas where the extra time spent upfront would be most valuable. Ultimately how to progress through the workflow and where to cut or spend extra time depends on the type of molecule.
I was particularly interested in the Ibex® Design offering delivering tox drug product in just 9 months. They also guarantee supply of at least 1.5 kg of GMP drug substance. This timeline also includes stability studies, storage and management of supply chain along with a manufacturing slot reserved for clinical resupply. At the end they supply submission-ready CMC data for IND or IMPD.
I found all of the talks interesting and very worthwhile for companies navigating their biotherapeutic from discovery through clinical trials and beyond.
To learn more, there will be another virtual symposium hosted by the MassBioHub on October 27th. To see the full agenda and to register, please see Approaches and tools for maximizing success on the journey from discovery to the clinic. Dr. Stallwood and Dr. Porter will be presenting along with other industry experts and local biotech companies.