

Ten Years of Induced Pluripotent Stem Cells: A Look Back
Authored by Debbie King, Contributor, Cell Culture Dish
This year, I had the opportunity to attend the International Society for Stem Cell Research (ISSCR) conference held in San Francisco, CA. It was wonderful to hear about the ‘hot topics’ in Stem Cell Research and to see new technologies from industry leaders. On the second day of ISSCR2016, I had the pleasure of hearing Shinya Yamanaka give a talk on induced pluripotent stem cells (iPSCs). The title of his talk was ‘Reprogramming of Cells and Scientist’. The first part was clearly autobiographical while the second half was focused on basic stem cell science (NAT1’s role in pluripotency). It was clear he was reflecting on how iPSC technology has changed him as a scientist as well as the scientific community as a whole. This year marks the ten year anniversary of the first induced pluripotent stem cell paper by Shinya Yamanaka and Kazutoshi Takahashi. He was awarded the 2012 Nobel Prize in medicine, which he shared with John B. Gurdon, for this revolutionary discovery.
What are Induced Pluripotent Stem Cells?
In brief, iPSCs are generated from adult cells by introducing specific pluripotent-associated genes to reprogram them to a pluripotent or ‘embroyonic stem cell-like’ state. The most well known pluirpotent stem cells are the embryonic stem cells (ESCs) derived from the inner cell mass of a blastocyst (pre-implantation stage embryos). ESCs and iPSCs have two key characteristics in common:
- They have the ability to proliferate continuously
- They can give rise to cell types from all three germ layers (endoderm, mesoderm and ectoderm)
These properties make pluripotent stem cells a powerful tool for a range of scientific endeavors ranging from study of basic cellular mechanisms to disease modeling, drug discovery and cell-based therapies. Figure 1 shows some highlights in the timeline of iPSC technology which will be described throughout this article.
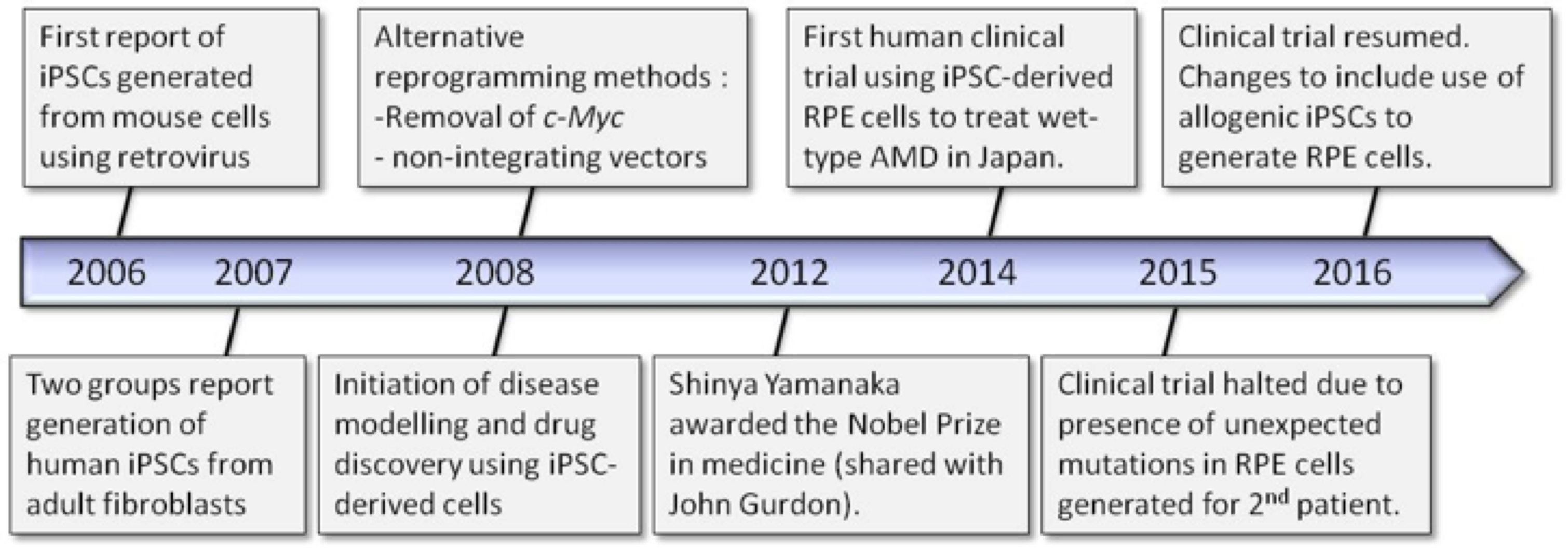
In 2006, Yamanaka and his team screened twenty-four genes thought to be important for pluripotency and introduced them into mouse fibroblast cells using retroviral delivery. They showed that just four of the twenty-four genes were required to generate mouse ESC-like colonies. The transcription factor genes used were Oct3/4 (octamer binding transcription factor 3/4), Sox2, Klf4 and c-Myc dubbed the “OSKM factors”. These mouse iPSCs showed unlimited self-renewal capabilities and demonstrated functional pluripotency through in vitro embryoid body (EB) formation and in vivo teratoma formation and fetal chimeras.
The generation of human iPSCs from adult fibroblasts followed quickly just one year later in 2007. Yamanaka et al successful reprogrammed adult human fibroblasts using the “OSKM factors” in a retroviral system and Thomson et al showed successful reprogramming with a different set of four factors: Oct3/4, Sox2, Nanog, Lin28 using a lentiviral system (Yu et al, 2007).
Labs around the world hurried to use this revolutionary technique and by late 2009, only 3 years after the original publication, some 300 papers on iPS cells had already been published (Scudellari, 2016). Human iPSCs have gained popularity over human ESCs because they circumvent the ethical controversy surrounding the destruction of embryos to establish human ESC lines. While the scientific community has flocked to use this technology, there have been challenges in reprogramming cells such as low efficiencies, genomic insertion of vectors, and the risk of tumorigenesis that needed to be overcome.
iPSC Hurdles
Many published reprogramming techniques show efficacy for reprogramming cells to an ESC-like state, but they are generally inefficient with only a small fraction of cells being fully reprogrammed. In Yamanaka’s original mouse study, the percentage of cells successfully reprogrammed ranged from 0.01 – 0.1% (Takahashi and Yamanaka, 2006). Therefore, many researchers have attempted to discover new molecules, which could facilitate reprogramming and increase its efficiency.
It is also known that some of the reprogramming genes, particularly c-Myc, are oncogenic, which brings about the potential risk of carcinogenesis, which is troubling for the prospect of iPS cells in clinical therapies. In an effort to address this, groups have published reprogramming protocols where c-Myc was omitted (Nakagawa et al, 2007) or replaced with alternative genes. The Thomson lab was able to successfully able to reprogram cells using Nanog in place of c-Myc (Yu et al, 2007).
In addition, the use of integrating viruses as a delivery system, while effective to reprogram cells into iPSCs, could lead to undesired insertional mutagenesis in the target cells. A common strategy to avoid genomic insertion has been to use non-integrating vectors such as adenovirus, Sendai virus, episomal plasmids and transposon vectors (Malik and Rao, 2013). Use of small molecules (Huangfu et al, 2008), protein compounds (Zhou et al, 2009) and microRNAs (Judson et al, 2009) to reprogram cells have also been successful.
It has been ten years since the first iPSCs were generated and the mechanism of reprogramming are still not well understood. Moreover, iPSCs retain ‘epigenetic memory’— a pattern of chemical marks on their DNA that reflects their original cell type from the reprogramming process. There is debate as to whether these differences are relevant in the context of cell-based therapy. And, as with all cell lines, iPS cells vary from one strain to another, which has made it difficult to establish controls in experiments. While safety concerns could impede iPSC-based therapies, iPSCs have been invaluable for disease modeling and drug discovery/screening.
Disease Modeling/Drug Screening & Discovery
Since the mammalian genome is highly conserved between species, modeling human disorders using animal models such as the mouse, rat and non-human primates has enabled the dissection of disease mechanisms at different developmental stages and in a variety of cell types in vivo. However, due to differences between the species acquired after ancestral divergence, not all human disease phenotypes can be recapitulated using these models. In these cases, it is preferable to use patient-derived cells that can be cultured to study a disease phenotype. For example, cancer studies often rely on tumor cells easily isolated from patients. Likewise, modeling of genetic disorders can be facilitated by using patient-derived immortalized cell lines created from blood or tissue biopsies. However, many disease phenotypes are manifested in cell types that are difficult to isolate from patients and/or cannot be sustained for more than a few passages in culture. These limitations can be overcome when using iPSCs making them attractive for disease modeling.
iPS cell lines have been generated for a wide variety of human diseases, including Duchenne (DMD) (Park et al, 2008), Down syndrome (Park et al, 2008) and polycystic kidney disease (Freedman et al, 2013). In many cases, the iPSC-derived from patients exhibiting a disease show cellular defects not observed in iPSCs from healthy patients, providing valuable insight into the pathophysiology of the disease (Grskovic, M et al, 2011).
The current outbreak of Zika virus (ZIKV) in South America and its potential spread worldwide has led to a global health emergency and there is urgency in the scientific community to understand the mechanism of action to aid in discovering potential treatment. Recently, Qian et al (2016) used iPSCs to make brain organoids (three dimensional ‘mini-brain’) to determine whether and how infection with ZIKV in pregnant women could lead to microcephaly in the developing fetus, a condition where the brain does not develop normally resulting in a smaller than normal head. When the organoids were exposed to the ZIKV, the virus was found to preferentially infect neural stem cell progenitors causing increased cell death and reduced proliferation, resulting in microcephaly in the fetus. These and other findings have helped the US Centers for Disease Control and Prevention (CDC) to conclude that ZIKV does in fact cause microcephaly.
Now, with the availability of new gene editing technology such as CRISPR/Cas9, researchers have also been able to introduce disease-associated mutations into a sample of iPS cells and then compare them back to the original, unedited cell lines thus allowing for the appropriate isogenic controls in experiments. This is critical to elucidate true molecular differences stemming from a particular disease as opposed to differences observed due to inherent variability in the iPSC lines themselves. iPSC technology combined with the power of CRISPR/Cas9 gene editing will lead to exciting advancements in our understanding of human disease.
iPSC technology has been impactful for drug discovery as well. The use of iPSC-derived disease cells can be used to screen or test experimental compounds for treatment. In 2012, for example, iPSCs derived from individuals with familial dysautonomia disease (rare, fatal genetic disorder affecting neural crest lineages) were differentiated into neural crest precursor cells. These cells were subsequently used to screen 6,912 small-molecule compounds to identify a potential drug to treat the condition (Lee et al, 2012).
The field of regenerative medicine is really where the huge potential of iPSC technology has yet to be realized. The ability to have personalized therapies to treat disease through generation of required cell types from reprogrammed patient cells would remove the risk of immune rejection, and sidestep the ethical concerns of using cells derived from embryos. However, the road to cellular therapies has been difficult and only one clinical trial using iPSCs has been conducted to date.
Clinical Landscape
To date, there has only been one clinical trial involving iPSC-derived cells to treat exudative (wet-type) macular degeneration (AMD). In wet-type AMD, there is an abnormal growth of new blood vessels behind the retina of the eye, which damages the retinal pigment epithelial (RPE) cells causing distorted vision or loss of visual clarity; a common affliction in the elderly. This study was conducted by Kobe-based RIKEN Center for Developmental Biology, led by Dr. Masayo Takahashi. In 2014, sheets of RPE cells, differentiated from patient-derived iPSCs, were transplanted into the right eye of a 70-year old female patient. The tranplants covered the retina to provide support to still-functioning photoreceptors. At the six month mark post-transplantation, the patient experienced no new, aberrant growth of blood vessels. Her visual acuity was stabilized and there have been no safety related concerns to date (Kagimoto, 2015).
The trial suffered a setback prior to the treating a second patient with their own iPSC-generated RPE cells. Mutations were detected in the RPE cells that were not present in the patient’s original fibroblast cells, including a mutation in a known oncogene. While further analysis showed the risk of carcinogenesis from these mutations was very low, this unexpected result combined with changing government regulations at that time in Japan led RIKEN and Takahashi to suspend the trial in 2015.
Finally, in June 2016, the Japan Times reported that the clinical trial will resume. RIKEN will work in collaboration with the Center for iPS Cell Research and Application (CiRA) in Kyoto, Japan, which has several well characterized iPSC lines that have been rigorously tested to meet strict safety guidelines. The choice to switch from autologous therapy used with the first clinical patient to allogeneic could be an important one when looking ahead to wide-level clinical therapy, allowing for mass-production and standardization. While patient-specific treatment is most ideal theoretically to avoid immune rejection, the process is time-consuming and costly (the Cell Therapy for one patient in the RPE cell trial cost $1 million and took one year to execute). This tradeoff of cost and efficacy will continue to be evaluated as more iPSC cell therapies are explored.
iPSCs: Looking Ahead
Developing and testing an iPSC therapy in even one person has proven to be a valuable ‘proof of principle’ endeavor. In the coming years, other iPSC-derived cellular therapies are expected to emerge in countries worldwide. Moreover, therapies derived from embryonic stem cells, and adult stem cells will likely help provide more guidelines for cellular therapies as a whole that will benefit the iPSC-based therapies.
Several groups are working towards iPSC Cell Therapy for Parkinson’s disease (PD). PD is characterized by the progressive loss of dopaminergic (DA) neurons in the substantia nigra region of the brain leading to tremors, rigidity, bradykinesia, and other debilitating symptoms. In Japan, Jun Takahashi with the CiRA institute aims to transplant iPSC-derived DA neural progenitor cells into PD patients’ brains to repopulate DA neurons lost because of the disease. The team is currently confirming efficacy and safety of the DA neural progenitors in non-human primate model before moving to human trials. At Scripps, in La Jolla CA, Jeanne Loring and her team just received $2.4 million in funding from the California Institute for Regenerative Medicine (CIRM) to support testing of iPSC-derived patient-specific Cell Therapy for PD as well.
For now, there is a recognized need to ensure safety of any iPSC-derived therapies and there is a push to systematically verifying cell lines’ identity and safety, by checking their genomes, gene-expression patterns and more (Scudellari, 2016). For example, the study by Bhutani et al (2016) was the first ever to evaluate the safety of three popular reprogramming methods (integrating retroviral vectors, non-integrating Sendai virus and synthetic mRNAs). Using whole-genome sequencing and de novo genome mapping to compare iPSCs to their parent fibroblasts, the researchers conclude the process of reprogramming is unlikely to introduce mutations that make the cells unsuitable for therapy. Harmful mutations could accumulate downstream, however, as the cells are cultured and subsequently differentiated thus highlighting again the importance of quality control for cell-based therapies. In March, the European Bank for Induced Pluripotent Stem Cells released its catalogue of standardized iPSCs for use in disease modeling. Similarly, Shinya Yamanaka is involved in banking iPSC lines that would be immunologically compatible across a broad population to support more allogeneic approaches. Articlesization and rigorous safety testing are going to help advance the use of cell therapies forward but will require a concerted effort from scientists, pharmaceutical industry and governing agencies to achieve.
The impact of iPSC technology on basic research, disease modeling, drug discovery and regenerative medicine has been monumental in such a short timeframe and undoubtedly there are many more exciting discoveries in the decades to come.
ISSCR Series
This is one in a series of blogs covering ISSCR 2016. Find out more about the ISSCR’s 2017 Annual Meeting in Boston, June 14-17 by visiting – ISSCR’s 2017 Annual Meeting
ISSCR Series Blogs
Cell Therapy for Parkinson’s Disease – An update on the move toward clinical trials
References
Bhutani K, et al. Whole-genome mutational burden analysis of three pluripotency induction methods. Nature communications, 2016; 7, Article number: 10536.
Freedman BS, et al. Reduced ciliary polycystin-2 in induced pluripotent stem cells from polycystic kidney disease patients with PKD1 mutations. Journal of the American Society of Nephrology 2013; 24 (10): 1571–86.
Grskovic M, et al. Induced pluripotent stem cells–opportunities for disease modeling and drug discovery. Nature reviews, Drug Discovery 2011; 10 (12): 915–29.
Huangfu D, et al. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nature Biotechnol., 2008; 26, 795 – 797.
Judson RL1, et al. Embryonic stem cell-specific microRNAs promote induced pluripotency. Nat Biotechnol, 2009; 27(5):459-61.
Kim K, et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010; 467, 285–290.
Kagimoto, H. The path forward: Letter to editor. Cell Gene Therapy Insights 2015; 1(1), 19-20.
Lee G, et al. Large-scale screening using familial dysautonomia induced pluripotent stem cells identifies compounds that rescue IKBKAP expression. Nature Biotechnol. 2012; 30, 1244–1248.
Malik N & Rao MS. A review of the methods for human iPSC derivation. Methods Mol Biol. 2013; 997: 23–33.
Nakagawa M, et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nature Biotechnol. 2007; 26, 101–106.
Park IH, et al. Disease-specific induced pluripotent stem cells. Cell 2008; 134 (5): 877–86.
Qian X, et al. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell 2016; 165, 1238–1254.
Scudellari J. How iPS cells changed the world. Nature, News Feature, 16 June 2016; 534, 310–312.
Takahashi K et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007; 131 (5): 861–872.
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006; 126 (4): 663–76.
Yu J, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007; 318 (5858): 1917–1920.
Zhou H, et al. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell, 2009; 4 (5): 381–4.