
There’s a New CAR in Town: CAR-Expressing Natural Killer Cells
The ability of the body’s immune cells to target and eliminate infectious organisms and foreign invaders has been studied for decades. It is no surprise that scientists want to harness the power of these cells in immunotherapy to specifically target and eliminate unwanted cells in the body. CAR (chimeric antigen receptor)-expressing T cell adoptive cell therapies have generated a lot of excitement and investment in recent years due to the unprecedented clinical results, which is driving research forward at a breakneck pace. The general steps for CAR-T are as follows: T cells are extracted from a patient’s blood, genetically modified with a chimeric antigen receptor specific to a surface antigen on target cells, culture-expanded and then reinfused back into the patient. To date, the U.S. Food and Drug Administration (FDA) has approved two CAR-T therapies (Novartis’ Kymriah® and Kite/Gilead’s Yescarta®) targeted against hematologic malignancies, and currently hundreds more clinical trials are underway for other malignancies, including solid tumors. However, the autologous (patient-specific) nature of this cell therapy, complex manufacturing workflows and the risk of graft versus host disease (GVHD) has raised concerns over its cost and safety. Similarly, obtaining enough lymphocytes from an ill, often lympho-depleted patient can pose a barrier to generating clinically relevant doses of CAR-T cells.
Because of these limitations, researchers are looking at other immune cells – namely, the natural killer (NK) cells – to modify with the same type of cancer-homing CAR receptor. NK cells are attractive contenders because they have potent anti-tumor activity, and their safety in an allogeneic, “off-the-shelf” format could overcome some of the challenges facing the current paradigm of autologous CAR-T cell therapies. Decreased production costs mean more available treatments to treat more patients in a shorter timeframe. Also, CAR-modified NK cells may provide some additional cancer-killing advantages over CAR-T cells, through their native receptors, and better tumor surveillance capabilities, which may make them more effective against certain malignancies where CAR-T cells have been less successful, like solid tumors1.
What are NK Cells?
Discovered in the early 1970s2, NK cells are large granular lymphocytes and potent cytolytic effectors of the innate immune system. Descended from the common lymphoid progenitor, which also gives rise to B and T lymphocytes and dendritic cells, NK cells are the body’s first line of defense against infections and tumor formation. Unlike T (and B) cells, NK cells are able to mount a rapid immune response against cancer and virus-infected cells without prior sensitization, which is how they became known as “natural” killers. NK cells achieve this by releasing cytotoxic proteins, primarily granzymes and perforin, stored within secretory lysosomes, which induce target cell lysis. They can also release a host of cytokines that recruit other immune cells to the site of infection3. Their ability to target cells that are missing the “self” MHC (major histocompatibility complex)-1 markers is unique because harmful cells that lack or downregulate their MHC1 markers go undetected by other immune cells (like T cells) 3.4. This HLA (human leukocyte antigen)-independent mechanism makes NK cells very attractive for cancer immunotherapy.
NK Cell Subsets
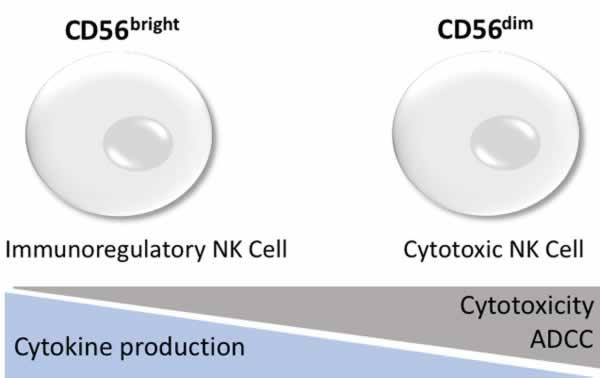
Human NK cells are characterized by the expression of CD16 (FcƔ receptor III), and CD56 and by their lack of T cell receptor (TCR) and CD3 expression. They are further subdivided based on their CD56 expression levels, CD56bright and CD56dim, which differ in their cytotoxic potential and cytokine production5. Evidence suggests that CD56bright NK cells are the precursor cells of the CD56dim subset, however, each population plays a relatively distinct and integral roles during the human NK immune response5,6.
NK Cell Activation and Target Recognition
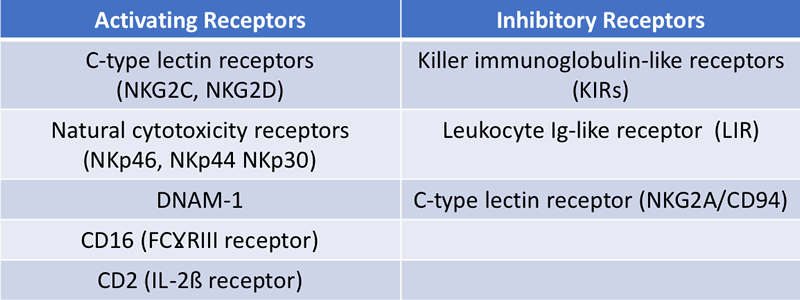
The NK cell’s HLA-independent specificity for target cells is not restricted by a single antigen receptor but is instead determined by the balance between activating and inhibitory surface receptors (Table 1). If the activating receptor signal is stronger than the inhibitory signal, target cell lysis is induced through by perforin and granzyme release. Conversely, when inhibitory signals are predominant, NK cytotoxicity is not activated and the cell is safe from destruction.
According to the ‘missing self’ hypothesis6, the function of NK cells is to recognize and eliminate cells that fail to express self-MHC-I molecules or express foreign MHC-1 (Figure 2). Additionally, when cellular stress or DNA damage occurs during viral infection or tumor formation, an upregulation of “stress ligands” on transformed cells results in activation of NK receptors inducing cytotoxicity. Cytokines important to NK activation include interleukin-12 (IL-12), IL-15, IL-18, IL-2, and CCL5 (Chemokine (C-C motif) ligand 5)5.
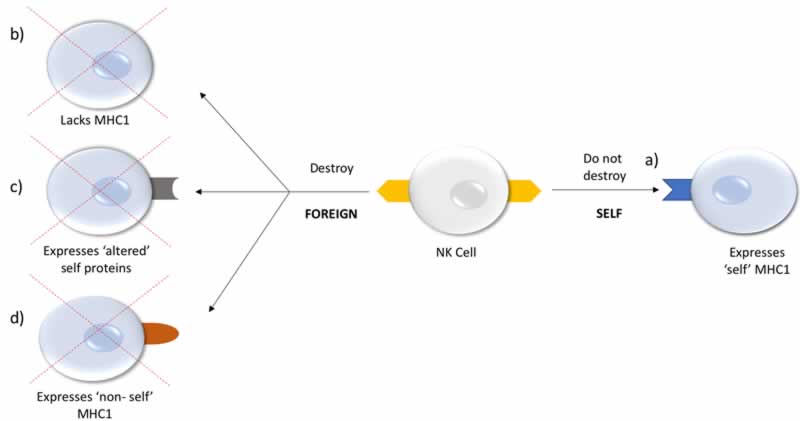
a) Self: NK cell recognizes self MHC1 expression on cell; no cell lysis.
b) Missing-self: Target cell has lost MHC1 expression on cell; NK cytotoxicity activated leading to cell lysis.
c) Stressed-self: Target cell expresses altered ‘self’ or stress-induced ligands; NK cytotoxicity activated leading to cell lysis.
d) Non-self: Target cell expresses foreign MHC1; NK cytotoxicity activated leading to cell lysis.
Sources of NK Cells for CAR-NK
Although autologous NK cells can be utilized for adoptive therapy, their in vivo efficacy has been limited, even with CAR engineering7. Autologous NK cells from cancer patients may have functional deficiencies making them unable to recognize malignant cells, and inhibition by self-HLA molecules may affect their persistence and therapeutic efficacy8. Allogeneic NK cells may be better for immunotherapy because they are not restricted by the patient’s HLA expression. Allogeneic NK cell sources include peripheral blood (PB), umbilical CB, bone marrow (BM), human embryonic stem cells (hESCs), induced pluripotent stem cells (iPSCs), or readily available NK cell lines7. Each has its own advantages and disadvantages in terms of scalability, transducibility and safety for CAR engineering. There is a plethora of information on the efficacy and safety of the various cell sources from studies done with unmodified NK cells for adoptive therapy, but CAR-modified NK cells represent a new paradigm that necessitates its own safety and efficacy testing.
Primary NK Cells
Primary NK cells can be obtained from both peripheral blood mononuclear cells (PBMCs) and umbilical cord (UC) blood cells. They can be difficult to isolate, purify, and transduce, often producing a heterogeneous cell population that expands poorly7. Commonly, magnetic antibody separation is used to isolate the NK population from other immune cells prior to genetic manipulation and expansion. CAR-transduced NK cells generated from frozen CB units obtained from large CB bank centers could improve the scalability of this cell source that is not achievable with individual adult donors who require screening and leukapheresis9.
Cell Lines
There are a number of cell lines derived from NK cells that are potential candidates for immunotherapies, including NK-92, NKL, KYHG-1, and NKG. These well-characterized, clonal cell populations with potent anti-tumor activity can be readily expanded to produce large numbers of CAR-transduced NK cells. The IL-2 dependent NK-92 cell line, derived from a patient with non-Hodgkin’s lymphoma, has shown higher cytotoxicity against a number of primary and established tumor cells compared to activated unmodified NK cells4. However, because of its cancerous origins, the NK-92 cells require irradiation before infusion into the patient to prevent their proliferation in vivo while maintaining cytotoxicity. Data from recent phase I clinical studies, where cancer patients were infused NK-92 cells, even up to doses of 1010 cells/m2, have proven to be safe7,10,11. Because of these results, CAR-engineered NK-92 cells are of great interest as a renewal source for clinical use.
Pluripotent stem cell-derived NK Cells
Human embryonic stem cells (hESCs) and induced pluripotent stem cells (iPSCs) can offer another renewable and potentially better source of NK cells. These undifferentiated cells can be culture expanded and subsequently differentiated into NK cells. However, finding the optimal hESC/iPSC cell line and differentiation protocol to obtain larger numbers of NK cells has yet to be achieved. That said, researchers have had success with iPSCs as a starting population for CAR-NK engineering. For example, clinical researchers, in conjunction with Fate Therapeutics, have developed a platform to generate NK cells from iPSC. In a 2-step differentiation protocol, hematopoietic CD34+ precursor cells are generated from hESC/iPSC embryoid bodies, which are further differentiated into mature NK cells after treatment with a specific cytokine cocktail10.
Both iPSC-derived NK cells and those from NK cell lines have the advantage that they can be extensively tested and characterized to maintain specific standards and offers the unique opportunity to manufacture CAR-NK cells under Good Manufacturing Practices (GMP) guidelines.
CAR Constructs
NK cells can be transduced to express chimeric antigen receptors (CARs) for cancer retargeting in much the same way as in T cells. The CAR constructs are composed of an extracellular antigen-recognition ScFv connected via a flexible linker to a transmembrane domain followed by an intracellular signaling/activation CD3ζ domain that provides a signal to activate NK cells.
CARs have evolved through the addition of costimulatory molecules to the intracellular CD3ζ to enhance cytotoxicity and durability. In the case of 1st generation CARs, a cluster of differentiation CD3ζ chain-derived signaling domain are responsible for effector cell activities while 2nd and 3rd generation CARs have one or two costimulatory domains, respectively, to boost CD3ζ function (Figure 3). In addition to engineered cancer-homing receptors, CAR-expressing NK cells retain their native NK cell receptor-dependent mechanisms, making them potent effectors for immunotherapy12.
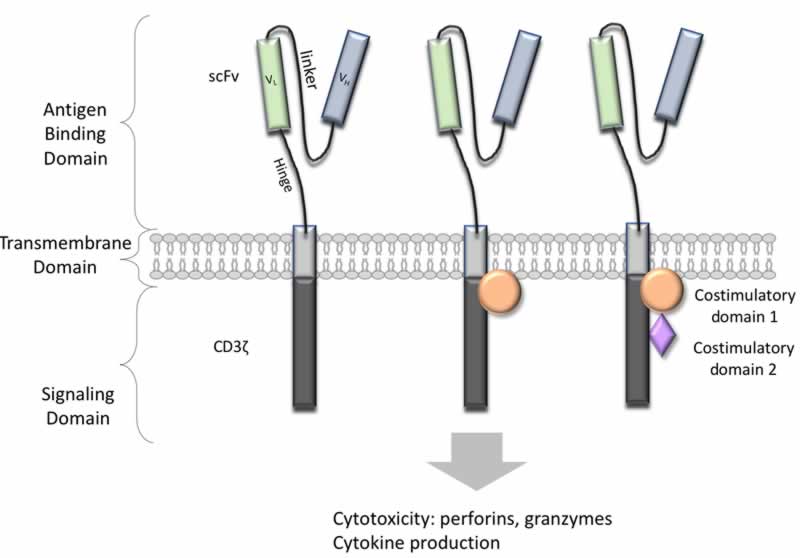
CARs can be transduced into NK cells via viral vectors and non-viral vectors. Both Ɣ-retrovirus- and lentivirus-based vectors have been used to transduce primary CAR-NK cells. The high transduction efficiency, between 43%–93%, is attractive but because of genome integration, there is risk of insertional mutagenesis12. In contrast, lentiviral vectors, while not as prone to insertional mutagenesis, are less desirable because their transduction efficiency is low.
On the other hand, non-viral vectors are inexpensive and easier to execute, but they cannot integrate into the genome for stable expression and stable gene transfer is required to enable sustained CAR expression in expanding and persisting effector cells. A non-viral Sleeping Beauty (SB) transposon system may be a promising alternative where stable transgene expression can be achieved without the risks associated with viral vectors12.
CAR-NK Clinical Trials
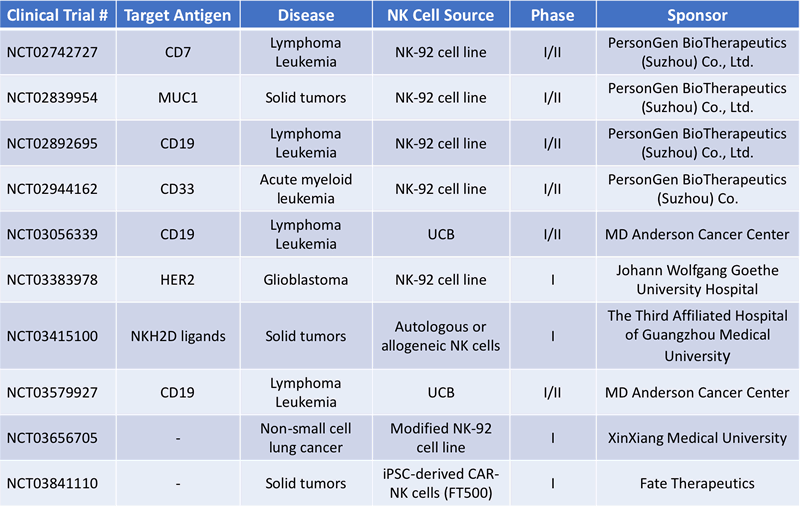
CAR-NK cells have two important advantages over CAR-T cells: safety and accessibility. CAR-T cells must be generated from the patient’s own T-cells to avoid GVHD. Because of this restriction, the manufacture of autologous T cell therapies requires long lead times and are extremely costly because one patient/donor can only generate one therapeutic dose. Allogeneic CAR-NK cells, on the other hand, don’t appear to cause GVHD and undesirable side effects are avoided by their short-term persistence and the lack of antigen clonality1. This could allow for an “off the shelf” product approach to greatly decrease time, cost and increase accessibility of CAR-NK therapy. Currently, there are a number of clinical trials currently underway involving CAR-NK cells for several indications (Table 2).
Leukemia and Lymphoma
In 2017, Dr. Rezvani, Dr. Shpall, clinical researchers at the MD Anderson Cancer Center (Houston, TX), initiated a phase I/II trial (NCT03056339) to treat patients with relapsed or refractory CD19+ B cell malignancies (chronic lymphocytic leukemia (CLL), acute lymphocytic leukemia (ALL), or non-Hodgkin lymphoma) using CAR-modified NK cells derived from primary, umbilical cord blood cells (UCB). This is a common malignancy that has had great clinical success with CAR-T cell immunotherapies.
UCB-derived NK cells are transduced with a retroviral vector expressing interleukin-15 (IL-15) and a CAR construct specific for the tumor-associated antigen (TAA) CD19 with the co-stimulatory domains of CD28 and the CD3ζ signaling domain. The construct also contains an inducible caspase-9-based suicide gene (iCasp9; iC9), which can be activated through a small-molecule dimerizer to induce apoptosis of the CAR NK cells in case adverse immune responses occurs13,14.
The group has initiated a second phase I/II clinical trial (NCT03579927) this year using the same CAR (CD19-CD28ζ2A-iCasp9-IL15)-transduced cord blood NK cells in conjunction with high-dose chemotherapy and stem cell transplant to treat patients with B-cell lymphoma.
Solid Tumor Malignancies
Dan Kaufman’s team from the University of California (UC), San Diego, chose a different starting source where they used iPSCs to generate NK cells for CAR transduction. The CAR construct consisted of the transmembrane domain of NKG2D, the 2B4 co-stimulatory domain, and the CD3ζ signaling domain to mediate strong antigen-specific NK cell signaling15,16. In a xenograph ovarian cancer murine model, mice treated with the iPSC-derived CAR-NK cells and those treated with CAR-T cells had reduced tumor growth after 21 days compared to control mice, which received unmodified NK cells. Additionally, the CAR-NK mice survived longer and did not suffer any ill effects such as weight loss, organ pathology, or increased cytokine levels compared with CAR-T treated mice, indicating that CAR-NK therapy might be a safer option than current CAR-T therapy, though more comparative species-specific studies need to be conducted17.
Kaufman, in collaboration with Fate Therapeutics gained FDA approval in November 2018 to use these iPSC-derived NK cells (dubbed FT500) in human clinical trials. In fact, in February 2019, 64 patients with advanced solid tumor cancers were enrolled in the phase I clinical trial15,1718 (NCT03841110) at the Moores Cancer Center in San Diego, CA. “This is a landmark accomplishment for the field of stem cell–based medicine and cancer immunotherapy. This clinical trial represents the first use of cells produced from human induced pluripotent stem cells to better treat and fight cancer,” says Dan Kaufman of UCSD in a press release.
Hurdles to Overcome
While the preclinical and clinical data thus far has been promising, there are still concerns surrounding CAR-NK cells that must be addressed, which is still in its infancy in comparison to CAR-T immunotherapy. For example, figuring out the best source of NK cells, improving ex vivo expansion, determining the optimal vector system, and designing CARs that work optimally for NK cells to provide the most potent therapeutic effect while maintain safety will be paramount7. Due to their short in vivo lifespan, NK cells are less likely to cause off-target effects as observed with CAR-T cells but their lack of persistence after adoptive transfer may limit the durability of their therapeutic effect12. Recent evidence suggest that the memory NK cell subpopulation may be better suited for CAR-NK adoptive therapy because they have a longer lifespan and in vivo persistence which could prolong their activity and durability to mitigate cancer relapse.
It seems unlikely that CAR-NK will replace CAR-T cell therapy in the near future because there are still many unknowns, but they could be a valued addition to the immunotherapy arsenal perhaps acting as a complementary therapeutic option to CAR-T cells. Because NK cells have an intrinsic cytotoxicity towards tumor cells that is independent of the CARs, CAR NK cells may be better at capturing tumor cells that have evolved mechanisms to evade the T cells4. Therefore, it may be possible that cancer clearance may be improved if both CARs are used in a combinatory approach than with CAR-T alone. There is more work to be done, but this is an exciting new direction for CAR-based immunotherapies.
Footnotes
-
1. Kloess S, Kretschmer A, Stahl L, Fricke S, Koehl U. CAR-Expressing Natural Killer Cells for Cancer Retargeting. Transfus Med Hemother 2019;46:4–13 doi.org/10.1159/000495771
-
2. Herberman RB, Nunn ME, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic acid allogeneic tumors. I. Distribution of reactivity and specificity. International Journal of Cancer 16(2):216-229, 1975.
-
3. Topham NJ, Hewitt EW. Natural killer cell cytotoxicity: how do they pull the trigger?. Immunology. 2009;128(1):7–15. doi:10.1111/j.1365-2567.2009.03123.x
-
4. Rezvani K, Rouce R, Liu E, Shpall E. Engineering Natural Killer Cells for Cancer Immunotherapy. Mol Ther. 2017;25(8):1769–1781. doi:10.1016/j.ymthe.2017.06.012
-
5. Boyiadzis M., Foon KA, Herberman RB. NK Cells in Cancer Immunotherapy: Three Decades of Discovery. Discovery Medicine. http://www.discoverymedicine.com/Michael-Boyiadzis/2009/07/28/nk-cells-in-cancer-immunotherapy-three-decades-of-discovery/ July 28, 2009. Accessed May 31, 2019.
-
6. Caligiuri M. A. Human natural killer cells. Blood. 2008; 112(3): 461-469. doi: 10.1182/blood-2007-09-077438.
-
7. Mehta RS, Rezvani K. Chimeric Antigen Receptor Expressing Natural Killer Cells for the Immunotherapy of Cancer. Front Immunol. 2018;9:283. doi:10.3389/fimmu.2018.00283
-
8. Geller MA, Miller JS. Use of allogeneic NK cells for cancer immunotherapy. Immunotherapy. 2011;3(12):1445–1459. doi:10.2217/imt.11.131
-
9. Liu E, Tong Y, Dotti G, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32(2):520–531. doi:10.1038/leu.2017.226
-
10. Dahlberg CI, Sarhan D, Chrobok M, Duru AD, Alici E. Natural Killer Cell-Based Therapies Targeting Cancer: Possible Strategies to Gain and Sustain Anti-Tumor Activity. Front Immunol. 2015;6:605. doi:10.3389/fimmu.2015.00605
-
11. Veluchamy JP, Kok N, van der Vliet HJ, Verheul HMW, de Gruijl TD, Spanholtz J. The Rise of Allogeneic Natural Killer Cells as a Platform for Cancer Immunotherapy: Recent Innovations and Future Developments. Front Immunol. 2017;8:631. doi:10.3389/fimmu.2017.00631
-
12. Hu Y, Tian ZG, Zhang C. Chimeric antigen receptor (CAR)-transduced natural killer cells in tumor immunotherapy. Acta Pharmacol Sin. 2018;39(2):167–176. doi:10.1038/aps.2017.125
-
13. Reno J. 'Natural Killer' Cells May Offer a Better Cancer Treatment Than CAR-T. Healthline: Health News. https://www.healthline.com/health-news/natural-killer-cells-may-offer-a-better-cancer-treatment-than-car-t#2 July 12, 2018. Accessed May 31, 2019.
-
14. Leslie M. Engineered natural killer cells may be the next great cancer immunotherapy. Science. https://www.sciencemag.org/news/2018/09/engineered-natural-killer-cells-may-be-next-great-cancer-immunotherapy September 13, 2018. Accessed May 31, 2019.
-
15. Knorr DA, Ni Z, Hermanson D, et al. Clinical-scale derivation of natural killer cells from human pluripotent stem cells for cancer therapy. Stem Cells Transl Med. 2013;2(4):274–283. doi:10.5966/sctm.2012-0084
-
16. Zhang J, Zheng H, Diao Y. Natural Killer Cells and Current Applications of Chimeric Antigen Receptor-Modified NK-92 Cells in Tumor Immunotherapy. Int J Mol Sci. 2019;20(2):317. doi:10.3390/ijms20020317
-
17. Natural killer cells for cancer immunotherapy: a new CAR is catching up. EBioMedicine. ;39:1–2. doi:10.1016/j.ebiom.2019.01.018
-
18. Zimmer K. Natural Killer Cells Prove Effective as a CAR Therapy in Mice. The Scientist. https://www.the-scientist.com/news-opinion/natural-killer-cells-prove-effective-as-a-car-therapy-in-mice--64456 July 6, 2018. Accessed May 31, 2019.