

Plasmid DNA Production for Cell and Gene Therapy
Company: MilliporeSigma
Job Title: Associate Director, EMEA Segment Development for Cell and Gene Therapy
High-quality plasmid DNA is a key component in cell and gene therapy manufacturing and as such is in high demand. This has led to the need to optimize manufacturing to meet the demand for volume as well as the quality required for use in the manufacture of therapeutics. Plasmid DNA (pDNA) manufacturing faces several challenges due to its large size, shear sensitivity, high viscosity, and the similarities between the pDNA and the impurities present during manufacturing. Thus, understanding all areas of the process are critical to successful manufacturing at a large scale.
With the high demand and quality requirements, it is important to seek out experts in the field who can illuminate the critical optimization opportunities for pDNA and answer questions about the production. For this Ask the Expert Session, we assembled a team of experts to answer questions on plasmid DNA production for cell and gene therapy applications.
Meet our Experts
 Nargisse El-Hajjami, PhD, Associate Director, EMEA Segment Development for Cell and Gene Therapy
Nargisse El-Hajjami, PhD, Associate Director, EMEA Segment Development for Cell and Gene Therapy
Molecular microbiologist and bioprocess expert with 10 years’ experience combining scientific research, process development, and biomanufacturing engineering. In her current role, Dr. Nargisse El Hajjami is focusing on cell and gene therapy business development by leading strategic initiatives, supporting go-to-market strategies, and supporting customers to build, grow and optimize their knowledge and manufacturing processes.
 Laurens Vergauwen, Process Development Scientist
Laurens Vergauwen, Process Development Scientist
Laurens is a downstream processing expert supporting customers in the development and optimization of various downstream purification technologies (chromatography, TFF, clarification, sterile and viral filtration). Being able to work with different kinds of manufactures, Laurens Vergauwen has gained a strong knowledge about purification strategies for different kinds of biopharmaceuticals, including plasmid DNA. Laurens holds a Masters Degree in Industrial Biochemical Engineering from the University of Leuven (Belgium) and his main passion is sharing knowledge.
This session is no longer taking submissions. To be notified of new Ask the Expert sessions, join our mailing list.
Question 1
Are there any key media ingredients that boost productivity?
Several parameters can affect the bacterial productivity like the master cell bank selection, the growth rate, the culture medium, the feeding rate as well as the appropriate growth conditions and parameters like pH, osmolarity, optical density (DO) and temperature.
The general media composition consists of a carbon source, a nitrogen source, magnesium sulfate, di- and monopotassium phosphate, and minerals. Both minimal and semi-defined media can be used. With minimal media, highly reproducible batches can be obtained. On the other hand, semi-defined media can support higher cell densities as the complex components (e.g. yeast extract) supply growth factors, amino acids, purines and pyrimidines.
As carbon source, glucose is conventionally used as it is efficiently metabolized and inexpensive. However, glucose may cause over production of acetate (Crabtree effect). This can be avoided by (partially) substituting glucose for glycerol. Glycerol can also aid in supporting reduced maximum specific growth rates.
Nitrogen is typically supplied by adding complex components such as yeast extract, casamino acids, or peptones.
Magnesium sulfate and potassium phosphates are added in order to provide a magnesium, sulfur, potassium and a phosphorous source (the phosphates also function as buffering agent). Other trace minerals are present in the complex components of the medium or can be added via specific trace mineral solutions.
The plasmid yield is largely affected by the C:N ratio. It is advised to test different ratios e.g. from 2:1 up to 8:1. The optimal ratio is different for each media type.
Question 2
My plasmid DNA is unstable, do you have any advice on what we could try to boost stability?
On one hand, it is important that the sequence of both your gene of interest (insert) and plasmid (vector) are well optimized. However, some plasmids will inherently be unstable. For example, if the insert is very large and/or contains inverted tandem repeats. For large genes, a small vector should be chosen. However, if this is already the case, it is likely that the plasmid will need to be produced with a low copy number (same in the case of inverted tandem repeats).
On the other hand, tweaking the growth conditions (e.g. growing at lower temperature), selecting an alternative host, optimizing the process conditions as well as choosing the right buffers for processing, formulating and storing your pDNA, can also help boosting its stability.
Also, DNAse contamination and pH greatly have great impact on pDNA stability. DNAse can degrade and digest the DNA double strands while extreme pH can break, denature and even change the pDNA sequence. Thus, selecting the right buffer and solution for your pDNA is fundamental for preserving the stability of your product for a long time. One of the best options is the Tris buffer with EDTA as the Tris buffer allows to control the pH to stabilize the pDNA, while the EDTA chelates inhibits DNAse activity.
For storage, the pDNA is usually stored at -20°C to - 80°C where it can stay stable for years, and it can also be stable at 4°C or at room temperature but for a short period.
Question 3
Why is GMP plasmid so expensive and in short supply?
GMP production is overall a more costly process. This has many reasons. The raw products used during GMP manufacturing are of higher purity, which comes at higher price tags. Dedicated production areas are required (e.g. cleanrooms). Validation is needed for cleaning methods and the final GMP product is of high purity, which has been confirmed via an array of validated analytical methods.
The short supply is due to the substantial growth of the global cell and gene therapy market in addition to vaccines and cancer treatment applications. In the last two years a big increase has been reported in the number of investigational new drugs and multiple drugs are being licensed for commercial distribution. Due to this increase, contract manufactures which provide GMP-grade plasmids are struggling to keep up with the increasing demand. The recent pandemic has exacerbated the problem, as a lot of vaccine candidates are being developed, including DNA-based vaccines and mRNA-based vaccine where the plasmid DNA is required as starting material for the in vitro transcription process of mRNA.
Question 4
How do I select the best plasmid for my gene?
There are two main types of vectors, cloning and expression vectors. Cloning vectors are ideal for the generation of many copies of your gene. If the goal is to express the gene of interest, an expression vector is needed.
For cloning vectors, the key aspects are the copy number (depending on the ori), selectable marker, and the cloning sites. Generally, a high copy number is preferred. Note that when the gene is toxic for the cell, or when the plasmid is unstable, a low copy number might be favorable. The selectable marker allows for the identification of a positive transformant. Most of the time these will be drug-resistance markers, but also auxotrophic markers are used. It is important to check whether the vector contains a cloning site which is suitable for the insert. To date, most vectors contain a multiple cloning site, which makes it likely that the vector is compatible with the selected restriction enzyme.
Expression vectors contain some additional sequences related to the expression, e.g., promotor, ribosome binding site, terminator, tag or fusion protein. Some of these sequences are specific to the host organism. Therefore, the expression vector needs to be compatible with the chosen host organism (e.g. mammalian, insect, E. coli).
Question 5
We are seeing very poor DNA yield. Do you have a good protocol?
In order to have a good plasmid DNA (pDNA) yield, the best protocol is to have a robust overall process where each step is well optimized. The pDNA purification is challenging due to its quite large size, high negative charge, viscosity and the fact that the contaminants have similar properties to the pDNA (open circular pDNA, genomic DNA, high molecular weight RNA). In addition, large plasmids can be sensitive to shear stress, further complicating the purification. In order to purify the supercoiled pDNA (desired form for therapeutics/transfection) with high yield, a well optimized downstream process is needed. Our company has solutions and capabilities for each step of the downstream process to assure an optimum pDNA yield and purity (summarized in figure 1). Considerations and watch outs for each downstream unit operations are discussed below.
[caption id="attachment_23053" align="aligncenter" width="800"]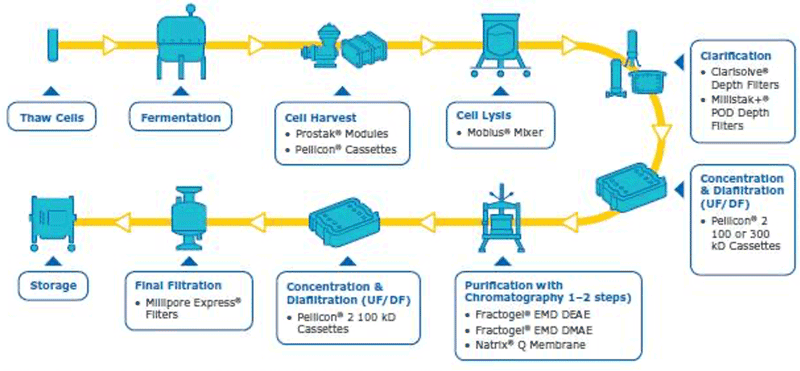 Figure 1: Integrated workflow with capabilities for pDNA purification from harvest to final fill[/caption]
Figure 1: Integrated workflow with capabilities for pDNA purification from harvest to final fill[/caption]
Cell harvest
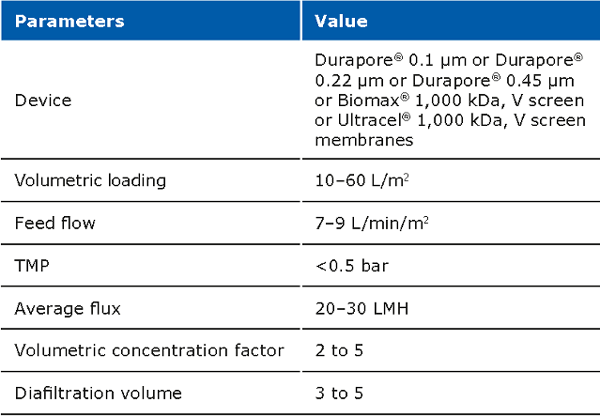
The cell harvest step uses centrifugation or microfiltration TFF. Centrifugation is often more cost effective when batch volumes (<10 L) or larger batch volumes (>1,000 L) need to be processed. When using centrifugation, special attention is needed for high shear generated in large scale centrifugation. Microfiltration can be performed with open-channel, flat-sheet TFF devices such as ProstakTM cassettes with Durapore® 0.1 or 0.2 μm microfiltration membranes or Pellicon® cassettes with Durapore® V screen or Biomax® 1,000 kD V screen ultrafiltration (UF) membranes. The open feed channels create a gentle flow path for cell retention, cause low shear, and can be used to process viscous and/or high solid feeds. When using membrane cut-offs such as these, it is important to utilize a two-pump (permeate-controlled) TFF system. The TFF harvest step typically involves a 2-5X volumetric concentration followed by a 3–5 volume diafiltration for washing out spent media components and extracellular impurities prior to further downstream purification. TFF harvest is typically operated at low transmembrane pressure (TMP; 3–5 psi) and ΔP (<7 psi) with a control on the permeate flux. Hollow fiber modules are also applicable, but may be accompanied with scaling issues due to the non-linear scalability.
Typical operating parameters for micro filtration TFF:
Cell Lysis
The methods used for cell lysis can be divided into two main categories – chemical (alkali, detergents, enzymes, osmotic shock) and physio mechanical (heat, shear, agitation, ultra-sonification, and freeze-thawing) lysis. Alkaline lysis (NaOH at pH ~12) accompanied by detergents such as sodium dodecyl sulfate (SDS) and Triton® X-100 is the most common approach. It is important to optimize the lysis incubation time as it directly impacts the quality and quantity of plasmid DNA. Longer incubation time could lead to irreversible denaturation of plasmid DNA and shear degradation of genomic DNA. It is critical to have efficient but not too aggressive mixing employed on the alkaline lysis step to ensure there are no pH extremes causing irreversible denaturing of the plasmids or degrading it due to excessive shear. Mobius® single-use mixers can be very effective for batch lysis.
During the alkaline lysis method, cells are treated at specific, narrow range of pH (typically around pH 12) at which the genomic DNA will be irreversibly denatured, while the pDNA double chain remains intact (pH range of 12.0 to 12.5). The optimum pH value varies depending on the type of plasmid and host strain. A deviation of more than 0.1 pH unit from the optimum value may affect the yield and it is therefore critical to maintain a tight control of the pH range during alkaline lysis. At a pH >12.5, pDNA becomes irreversibly denatured and if the pH is too low, genomic DNA won’t be completely denatured and could complicate further downstream purification process. The incubation time for a standard alkaline lysis is fairly short and the step is usually completed typically within 5 minutes.
After the alkaline lysis, the pH is neutralized. This is typically carried out by addition of a high-salt buffer, such as sodium or potassium acetate at concentration of 0.7M – 3.0M and pH ~5–7.5, with/without 1.0–1.5% CaCl2 (promotes RNA precipitation) in the presence of a detergent (0.2 - 1% SDS). Polyethylene glycol (PEG) and polyethylenimine can also be added to the neutralization buffer to promote the precipitation of genomic DNA. Homogenous mixing during neutralization and precipitation is critical to maintain pDNA quality.
Clarification
Clarification unit operations for pDNA processes should enable removal of solid content from the feed stream. Clarification can be performed via centrifugation and/or normal flow filtration. Traditionally, centrifugation is used for the clarification. When using centrifugation, one should pay attention to the shear stress, which may damage the structure of the supercoiled plasmid, resulting in lower yields. Centrifugation may also require a secondary clarification step. Clarification using normal flow filtration is the modern approach. When using this method, feed streams can be used which are either untreated, pretreated or preclarified. Pretreatment has a major impact on the clarification filter capacity and must be selected carefully along with a consideration of the scalability of the process. Pretreatment options include use of gravity settling and separation, Polygard® CR 1 µm/Polygard® CR 50 µm, bag filters stainless steel screen filters, paper filters, and centrifugation. The capacity of the Polygard® CR filters is typically in the range of 0.55–8 L/inch.
The recommended filter set ups for the clarification step are:

The expected capacity of the filters is strongly dependent on whether a pretreatment is performed:
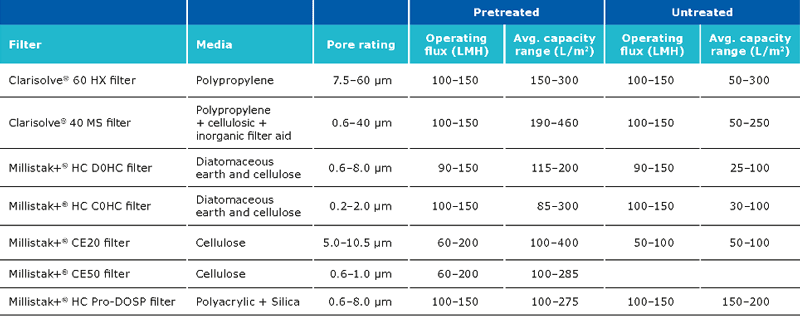
The typical recovery when using Clarisolve® and Millistak+® filters is >90%. The recovery can be increased using a chase wit salt containing buffer. The capacity of the Millipore Express® SHC sterilizing grade filter post clarification is generally between 400-650 L/m2.
Tangential flow filtration (TFF)
After clarification, there can be opted for a TFF step. This will allow to concentrate and perform diafiltration to exchange the medium into a buffer suitable for the downstream chromatography step. By performing a concentration prior to chromatography, one can reduce the chromatography loading time. In addition, RNA, small sized genomic DNA, and small proteins can be removed during TFF, which also helps prevent fouling of chromatography resins.
When performing TFF, some key watch outs are:
- Shear sensitivity of plasmids, careful adjustment of process parameters is needed to prevent damage to the plasmid.
- Membrane fouling, which results into yield loss.
- High salt buffers promote plasmid compaction, which may allow passage through small pores, resulting in yield loss.
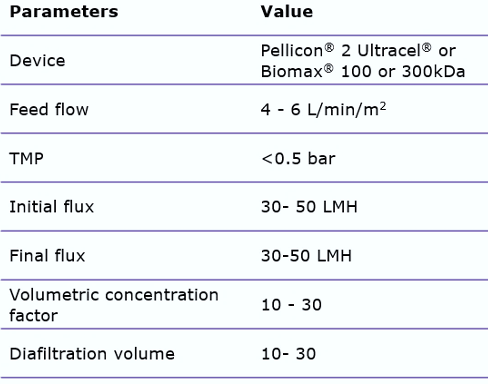
These issues can be avoided by carefully screening recommended open channel membranes, such as the Pellicon® 2 Ultracel® or Biomax® 100 or 300 kDa C-screen. By using open channels, shear stress can be reduced. In addition, minimizing the process time by appropriate membrane sizing is important. When using these wider membrane cut-offs, it is important to control the permeate flux, by utilizing a two-pump (permeate-controlled) TFF system. This will help prevent membrane fouling.
Typical TFF process parameters are:
Chromatography purification
For chromatography purification, typically anion exchange chromatography (AEC) and hydrophobic interaction chromatography (HIC) are used. Both techniques have been implemented for capture or intermediate purification/polishing and are often combined.
When using AEX as capture step for pDNA, it is beneficial to spike the clarified lysate with NaCl (120-250 mM). This will eliminate the interference of RNA, therefore increasing the binding capacity for pDNA. The optimal salt concentration for supplementation needs to be pre-determined, for example via a batch assay in microtiter plate format, measuring plasmid binding capacity at increasing sodium chloride concentrations. One can do this for different types of resins/membrane adsorbers. The following graph demonstrates this principle for the Fractogel® EMD DEAE (M) and Fractogel® EMD DMAE (M) resins:
[caption id="attachment_23056" align="aligncenter" width="500"]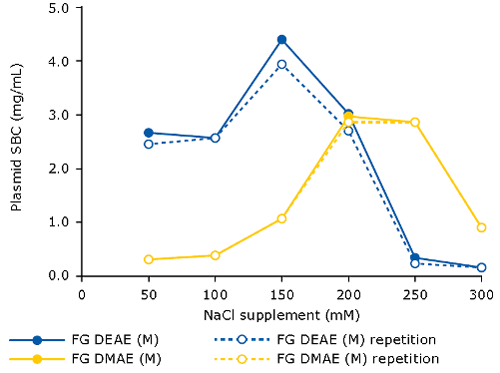 (original lysate before supplementation: pH 5.0, 67 mS/cm)[/caption]
(original lysate before supplementation: pH 5.0, 67 mS/cm)[/caption]
Recommended resins for plasmid purification and their respective performance:

Due to the large size of the plasmid, typically low binding capacities are observed on commercially available resins. This is due to the fact that most AEX media were originally designed for protein purification, as plasmid molecules are much bigger than proteins, they cannot enter the small pores, causing low binding capacity and slow mass transfer. Because the Fractogel® and Eshmuno® resins contain tentacle technology, typically higher binding capacities compared to other available resins can be achieved. The Natrix® Q is based on membrane technology and contains large convective pores. The latter promotes mass transfer and improves binding capacities (5-10 mg/mL). Due to the membrane technology, very short residence times are needed (<0.2 min). Because of these advantages, together with its single use format, it has been widely adopted.
All four resins mentioned in the above table can be used for the capture of pDNA. However, only the Fractogel® EMD DEAE (M) and Fractogel® EMD DMAE (M) resins are well suited for intermediate purification or polishing of plasmid DNA due to their moderate binding capacity and flow as well as good resolution due to a medium bead size (d50: 48–60 μm), clearing residual impurities like RNA and endotoxin efficiently.
Examples of HIC resins are Capto™ PlasmidSelect and Toyopearl® Butyl. The HIC step can either be located before or after the AEX step. When the HIC step is the first chromatography step, the eluate can directly be processed onto Fractogel® resins, as pDNA binding to these AEX resins tolerate the presence of elevated concentration of ammonium sulfate (pDNA binding capacity is not negatively affected).
Example of an experimental run for the Natrix® Q chromatography membrane for capture of pDNA:
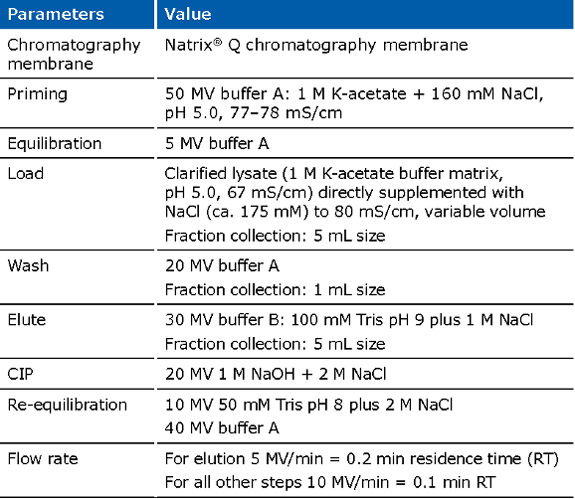
Final concentration and sterilizing grade filtration
After chromatography, the sample is concentrated and diafiltered in the buffer of choice. The same conditions apply as mentioned in the tangential flow filtration section.
The sterile filtration of pDNA can be challenging due to the large size and viscosity of the final product. In addition, large plasmids can be shear sensitive and may be damaged during the operation. Some key considerations are:
- Salt concentration: plasmid DNA tends to be more compact when increasing the salt concentration. The latter can lead to an improved yield and filter capacity.
- Membrane type: typically, a higher filter capacity and flux are achieved when using PES based membranes. PES based membranes can also be less damaging to larger plasmids (less shear). The Millipore Express® SHC is recommended for this operation.
- Plasmid purity: supercoiled plasmids tend to give better filtration performance compared to open-circular plasmids. Therefore, both yield and filter capacity tend to increase with purity.
- Defining the filtration endpoint: finding suggest that membrane fouling is correlated to yield loss. Optimizing the filtration endpoint can lead to an increase in yield.
- Feed flux or pressure do not show a significant impact on filtration capacity or yield. However, high driving forces should be avoided to lower potential shear stress.
Expected performance for the sterilizing grade filtration of purified pDNA:
Question 6
What kind of testing should we be doing to evaluate our plasmid DNA quality and in what ways can quality affect vector manufacturing
The quality and purity of plasmid DNA is of utmost importance for a successful transfection. Key watch outs are phenol, endotoxins and sodium chloride. Phenol and endotoxins can be harmful for the cells and salts can interfere with lipid complexing, which results in a decrease in transfection efficiency.
In order to determine the quality, one could assess (but not limited to):
- DNA concentration (OD 260 nm)
- DNA purity (OD 260/280 nm or UV-scan)
- Endotoxin (LAL test)
- Osmolality
- Residual genomic DNA (agarose gel electrophoreses or qPCR)
- Residual RNA (agarose gel electrophoresis)
- pH
- ccc monomer content (HPLC or via agarose gel electrophoresis)
With our BioReliance testing services, we offer a wide range of testing to helpyou assess the quality of your plasmid DNA that goes from Biosafety testing requirements (Sterility, Endotoxin, Host Cell Protein, Host Cell DNA, Sequencing, Plasmid conformation) to Product Characterization tests (Molecular Weight (ID) with Mass Spectrometry (MS), Purity – SEC/IEX/RP (depending on pDNA size), Concentration – A260/280)
Question 7
We are removing plasmid by using a benzonase treatment. Is this what you would recommend or are there better solutions?
The removal of DNA is one of the key challenges for viral vector manufacturing. In order to achieve the regulatory requirements of <10 ng DNA/dose and DNA size <200 bp, a good strategy is needed. A typical strategy is composed out of at least three different technologies:
- DNA-digestion, using endonucleases
- Tangential flow filtration in order to remove fragmented DNA
- Chromatography purification
Currently, Benzonase® endonuclease is seen as the industry standard for DNA digestion in vector processing. It is an efficient endonuclease which can degrade all forms of RNA and DNA (both single and double stranded). One unit of Benzonase® endonuclease is able to degrade approximately 37 µg DNA in 30 min to as low as 3-8 base pairs. Benzonase® endonuclease is provided in three different quality grades in order to meet the widest possible range of processing and cost requirements. The highest purity grade for example is the Benzonase® endonuclease Safety Plus Emprove® Expert, which is GMP manufactured and animal origin free. As it is part of the Emprove® program, it is backed up by quality dossiers, which can help you fast track through regulatory challenges.
In order to optimize the use of Benzonase® endonuclease, it is advised to perform a small DoE experiment. The three parameters that need to be examined are concentration, incubation time, and temperature. An example of a DoE set up can be found here. It is also preferred to examine at which step in the process the use of Benzonase® endonuclease is optimal, e.g. before cell lysis, post cell lyses or after a clarification unit.
Digestion of DNA using endonucleases has several benefits:
- Limits virus-nucleic acid complexes (makes purification unpredictable due to shift in pI and/or retention time), therefore increasing the yield.
- Protects downstream equipment against DNA fouling
- Reduces the viscosity
General information on Benzonase® endonuclease can be found here.
Question 8
Can you review the options for purification of plasmid DNA with your advice on which to pursue?
The different options and recommendations for the purification of plasmid DNA are discussed and developed above in question 5. For more information and data about the optimum parameters for the pDNA purification with our capabilities, you can refer to our white paper about “Scalable purification of Plasmid DNA: Strategies and considerations for Vaccine Manufacturing”.
Question 9
How much starting material is required for production?
In regards to plasmid production, an optimized fermentation process with cell densities ranging from 40-60 g/L can achieve pDNA yields around 1-2 g/L when a high-copy number plasmid is used.
For viral vector production, 0.5 mg of pDNA is required for transient transfection of viral vector (AAV or lentivirus) per one 1 L bioreactor. 1 L of transfected bioreactor typically yields 1E14 viral genomes for AAV and 3E9 viral genomes for lentivirus.
Question 10
When do you need to invest in GMP plasmid and when is research grade ok?
For the production of viral vectors, it is increasingly likely that the FDA regulations will soon recommend the use of GMP compliant plasmids for clinical batches. Plasmids are a critical material attribute, as it can impact the safety and quality of the final product. Therefore, it is highly recommended to start using GMP compliant plasmids when producing clinical batches (starting from phase 1). In addition, one will be prepared once regulatory bodies adjust the regulation. For process development and pre-clinical batches, research-grade plasmids can be used.
For NA-based vaccines, cancer therapies or gene therapies where the pDNA is injected directly into a human body, GMP grade plasmids are required.
Question 11
How can we reduce the cost of our viral vector manufacturing, specifically our plasmid DNA costs?
Several parameters can affect the costs for viral vector manufacturing, such as the choice of cell line (adherent versus suspension) and the process strategy (single-use versus multi-use solutions). Data suggest that the upstream costs can be lowered by up to 57% when opting for suspension culture versus adherent cultures. Single-use solutions eliminate the need for cleaning and cleaning validation and allow for higher flexibility and increased manufacturing capacity.
Cost reduction for pDNA manufacturing specifically, can be realized by efficient downstream processing and the incorporation of single-use solutions. Product recommendations to obtain a high yield during downstream processing are discussed in question 5.
Question 12
Do you recommend outsourcing or manufacturing plasmid DNA internally?
Today, most if not all pDNA manufacturing is being outsourced to specialized manufacturers and CMOs. With the increasing demand of pDNA within the biopharma industry, demand for appropriate manufacturing capacity and expertise is also increasing. This rises the need to build long-term solutions to be ready to cover future market needs and ensure supply integrity by investing in inhouse pDNA manufacturing.
The cost of outsourcing is quite high, requiring highly specialized expertise and manufacturing facility. At the same time, building a pDNA production facility with the required expertise and capabilities is also costly investment. Both options have pros and cons and making decision about manufacturing your plasmid DNA in house or outsourcing it would require a strong cost assessment study for each option taking into consideration the availability of the required expertise and facility in alignment with the company short- and long-term vision.
For example, a company using pDNA as raw material for viral vector production or mRNA synthesis would have a different vision and need compared to a company using pDNA as a final product for vaccine or direct gene transfer therapy. In other words, outsourcing the core activity of a company is different from outsourcing part of the process.
Outsourcing enables to benefit from required competencies and capabilities along with quality, efficiency, productivity and speeding time-to market. However, it is important to carefully select the partner based on the experience they have on the field, quality management systems, timelines and successful regulatory inspection history. Outsourced work is an extension of your business, so it is important to find the right partner with similar values as your own company.