

Biologic Products DNA to IND Timeline in 9 Months – Yes it can be done!
The ability to rapidly develop biologic products from conception to human clinical trials is an increasingly important aspect of controlling drug development costs and in expediting the drug’s pathway to one day provide critical treatments. Hence, there has been an increased focus on ways to shorten the timeline through product development. These expedited timelines are especially important when dealing with infectious disease control.
At the recent CBI Speed to IND for Biologics Conference in San Francisco, there were many interesting talks on strategies for reducing time to IND. One of the talks was a perfect fit as it discussed a process that brought a product from DNA to IND in a record 9-month timeline for for biologics drug development (in this case, recombinant protein therapeutics produced from mammalian cell culture). It also highlighted the kind of collaboration that was necessary to achieve such timeline. The talk, “From DNA to IND in 9 Months – How organizations worked together to meet a national healthcare initiative,” was presented by Jie Chen, Vice President of CMC Management at WuXi Biologics.
Meeting a National Healthcare Initiative
Dr. Chen began her talk by discussing how the standard timeline from lead selection to IND for most biologic products is more than two years. However, this timeline is not fast enough to combat emerging infectious diseases where every day can be critical to public health. She then explained that this was the situation with Tyzivumab, a monoclonal antibody against Zika infection. Tychan, the company who developed Tyzivumab, had a goal of getting the product into clinical trials in under a year to meet pressing public health needs. The product was considered a critical Singapore national infectious disease initiative. Even though Tychan had developed a unique drug discovery selection system to allow rapid drug product lead molecule identification, they needed a CDMO partner that could execute all aspects of the development from identified lead molecule to GMP manufacture of clinical product in order to meet their accelerated clinical trial timeline.
Tychan selected WuXi Biologics as their CDMO partner. Thus, Tychan and WuXi Biologics, along with input from Health Sciences Authority Singapore, Singapore-MIT Alliance for Research and Technology, Duke-National University of Singapore and National Research Foundation Singapore, created a collaboration to meet the goal of getting the monoclonal antibody into clinical trials in under a year for infectious disease control.
Keys to Reducing the Timeline
Dr. Chen explained that WuXi Biologics has a standard timeline of 18 months and an accelerated timeline of 15 months to bring antibody drug candidates from DNA to an IND filing. In the case of the Tychan program, six additional months needed to be shaved off from the 15M accelerated timeline in order to meet the project goal. Jie explained that one critical aspect to meeting this accelerated timeline was WuXi Biologics’s single-source integrated service model in which all development tasks could be done in-house without any transition gap to other parties to ensure the fastest turnaround time possible (Figure 1).
With this single-source approach, WuXi Biologics is able to complete all tasks from R&D protein generation to regulatory dossiers preparation internally, which enables a highly efficient product development timeline. In addition, the company’s effective project management eliminates common gaps found in traditional drug development using a multi-company/multi-vendor approach (e.g., segmented project stage transition, loss of material/knowledge transfer, and the time-consuming hand-offs between different companies, partners, and vendors).
The WuXi Biologics one-stop platform was established through experience with over 200 CMC projects from diverse biological molecules that included monoclonal and bispecific antibodies, fusion proteins, bioconjugates and other recombinant proteins. Dr. Chen emphasized the importance of a well-defined and characterized CMC platform. This means that the cell line development and cell culture process platform requires minimal optimization to achieve acceptable productivity and desired product quality. WuXi Biologics’ downstream and formulation development platforms allow quick evaluation of these functions to obtain acceptable process yield and Product Quality Attributes (PQAs). Other keys of the company’s CMC platforms include having multiple investigational tools to quickly resolve technical challenges and extensive analytical capabilities that permit streamlined assay development for in-process, release, stability, characterization and cell-based assays, and where appropriate assay qualification. Lastly, with PD, GMP cell bank construction and cell line characterization, GMP DS and DP manufacturing, stability and release testing in the same facility, or within very close proximity, the transition timeline gap for different CMC activities can be kept at minimum.
Next, Jie explained that the choice of areas where time savings could be achieved had to be carefully identified and a strategy needed to be established for implementation with appropriate risk control. Jie addressed that an important part of the strategy was the close working relationship with Tychan and its other collaborators. All involved parties worked together to support the fast track strategy to ensure the lowest risks associated with product quality and regulatory filing.
Risk Assessment and Mitigation
When cutting the timeline this aggressively, there are obvious risks that need to be assessed and mitigated. What I thought was most impressive was the way that WuXi Biologics leveraged the success and proven track record of their existing antibody technology platforms to confidently take these risks. They are able to utilize the knowledge of their platform to select the areas where time could be cut with minimal regulatory risks. For example, in cell line development they adopted a high-throughput and non-stop/parallel work flow strategy (i.e., no stopping to review results between the various CLD workflows). Hence, once stable pools emerged from multiple transfection events, they would move to cloning stage work without first conducting top pool selection. Similarly, later in CLD, they skipped the Pre-Master Cell Bank (PCB) generation and more than one Master Cell Bank (MCB) was constructed without waiting for final clone selection. The WuXi Biologics team also allowed 15 generations less cell line stability work to prove cell line stability from vial thaw to a scaled-up commercial manufacturing run, which helped to further shorten the timeline.
However, the aforementioned time-saving strategy comes with risks and they had to carefully assess those risks. For example, during cell line development, the top two clones selected for MCB generation were selected without waiting for bioreactor and product stability performance data. While this saved significant time, there was the risk that the eventual preferred clone or product would not have the desired properties in the subsequent scaled-up manufacturing process. So they had a mitigation plan to use ambr® analysis to evaluate clone performance in various simulated bioreactor conditions in parallel during clone selection. This is also where they could rely on the past data and success of their cell line development platform for early prediction of cell line stability and performance.
Another challenge was that in order to meet the timeline, there was not much time for process optimization, which created the risk of using a process that would generate a low product titer and yield and undesired impurity profile. Again, the WuXi Biologics team was able to leverage the knowledge of their CMC platform and the use of their high-throughput screening tools, to obtain process conditions faster and earlier. However, these high-throughput tools, which under this timeline ran in parallel across different functional areas (e.g., cell culture, formulation, and downstream development), generated a significant number of analytical samples that had to be analyzed in a short period of time. This could have caused a bottleneck for process development work but was resolved by increasing resources.
Another challenge for the team was the requirement of generating enough representative material throughout the program for process and formulation development work, assay qualification and toxicology studies. As several of these activities were being conducted in parallel and in order to meet the accelerated timelines, it was not possible to wait until cell line development was completed to use the final clone to generate materials needed for all of these activities. This strategy poses some risk as the material generated may not be completely representative of the same material generated from large-scale GMP manufacturing run, which used vials from the final selected MCB. Here they used for development studies, their proprietary WuXian protein expression technology to provide sufficient yet representative amounts of product. They had confidence from past programs that the material generated using the WuXian platform would be representative. A 10-liter confirmation production run using the final clone was then provided for stability and release assay qualification activities and the formal IND-enabling toxicology study instead of waiting for material from a larger scale (e.g., 200 L) non-GMP pilot run. The toxicology study for infectious disease only requires three months, which aligned with the remaining CMC activities (e.g., pilot and GMP manufacturing and release testing) so that the IND could be filed per the requested timeline.
Lastly, with a one-stop service offering, the WuXi Biologics team was able to make the most efficient transition flow from one activity to another with minimal time gap between each activity. They also employed the strategy of initiating process transfer to the GMP facility in parallel with later-stage PD activities. Having a robust and high quality GMP operation, they moved forward with using the MCB in GMP production with a conditional release. Likewise, once Drug Substance (DS) was generated they moved to Drug Product (DP) manufacture using a similar conditional release strategy without waiting for all assay lot release results to be finalized. In addition, because DS and DP manufacturing facilities are co-located in the same site, no time was lost moving material from one facility or to another vendor, which would have extended the timelines.
Tychan has also provided strong support at every decision point to ensure project progression without communication gaps. Tychan also discussed and received permission from the Singapore Health Authority to obtain an IND rolling submission. For example, they allowed the required one month drug product stability data to be provided in a supplement to the IND filing. WuXi Biologics added additional value to meet the IND timeline due to their large Regulatory Affairs (RA) team that was able to write the entire CMC package of the IND for Tychan. To keep from having the CMC package be the bottleneck at the close of the project, the RA team wrote each CMC section as it was completed thus streamlining the generation of the final packet.
Project Data
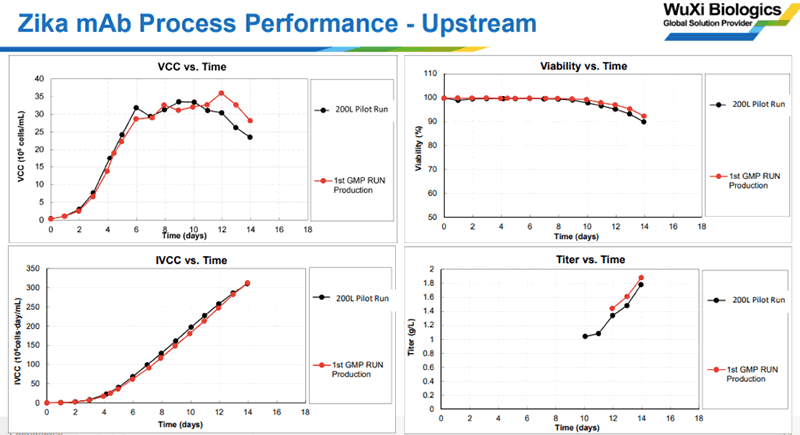
Jie presented data on both upstream and downstream process performance. The data showed excellent productivity and comparability between the pilot and manufacturing runs (as shown in Figure 2).
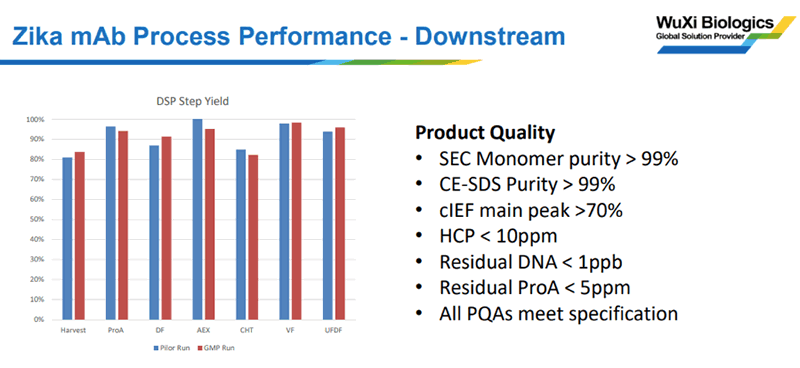
Downstream performance data demonstrated excellent product quality and comparability with the pilot runs (as shown in Figure 3).
Summary
In closing, Jie, summarized some of the key components in making this remarkable timeline work:
- Continuous cell line development activities without stopping for data review from transfection to final clone selection
- Clone stability data with 45 generations by skipping Pre-Master Cell Bank creation to achieve timeline reduction without compromising required cell line stability length
- Use of high-throughput systems to screen process parameters and reach process condition lock faster
- Creation of 2 master cell banks in parallel instead of waiting on final clone selection
- Use of platform process and methods for all functions as much as possible (USP/DSP/FFF/Assays)
- Dedication of more resources to handle all processes in parallel and to complete bolus of analytical samples generated by the parallel activities
- Initiate tech transfer earlier between process development and GMP site
- Prepare, procure and release raw materials earlier
- QA/QC system support to allow conditional release of the MCB for GMP use and conditional release of drug substance for drug product fill finish.
- Utilize WuXi Biologics experienced and dedicated regulatory team to prepare IND dossier as various CMC development activities completed to provide no time delay
- Effective Communications among all parties to align strategy ahead of time and if necessary to make a decision right on the spot!
Question and Answer
We were fortunate to be able to interview Jie Chen after her talk. Below is the transcript of our conversation.
- This work is so impressive; do you think it can be used to speed other infectious disease candidates to clinical trials?
Absolutely. In order to fight and control infectious disease outbreak quickly, our industry has to mobilize quickly to create a system which allows a fast timeline from target to drug product in clinic and that system has to include a regulatory authority playing a very active role. Exact timeline may vary from case-to-case since multiple risk factors must be addressed to know if that product’s characteristics and eventual clinical plan are appropriate for an expedited development strategy. A very well-established CMC development platform from lead identification to writing of the CMC package and a single-source contract service are the key for “plug and play” to make a timeline like this possible. Lastly, the manpower resources must be there and commitment by all parties involved is paramount to make this work, in addition to the extremely effective project communication plan and system.
- Is this something you would consider for more traditional biologics or is it better reserved for products where timelines are critical to public health?
Shortening drug discovery and development timelines is one of the most important goals for the Biopharmaceutical industry. At the conference, every speaker presented their efforts and strategies to reduce timeline to IND and to bring drugs to benefit patients faster. However, faster timelines cannot mean compromising on quality. As stated throughout my presentation, risks had to be addressed and mitigated to ensure product safety and efficacy. Otherwise, if there are complications the delays to fix and sort those out could be anywhere from a couple of weeks to many months. As a result, timing of the large-scale manufacturing runs, toxicology and clinical studies and even management, board and investor expectations are impacted greatly by any delays. When achieving a 9-month accelerated timeline, we worked hard and smartly to keep the risks low. We conducted whole cell line development from transfection to MCB and ensured cell line stability met cell banking and product commercial scale generation requirements. Both the upstream and downstream process used commercially available and quality raw materials and process conditions were developed and optimized to meet product release specifications. When conducting MCB and drug substance conditional release, we ensured safety-related testing data were available to avoid any safety concerns for GMP facility, and comprehensive risk assessments were performed and documented to meet regulatory guidance compliance. In summary, efforts of shortening the development timeline should be applied to all biopharmaceutical products, but the risks of each program may be different and must be weighed on a case-by-case basis. Therefore, a well-established risk evaluation and mitigation strategy needs to be established to bring project to success within its own achievable timeline.
- What was the biggest challenge during the process?
During early development time, in order to achieve the aggressive timeline, many development activities needed to be run continuously and in parallel. Data turnaround time needed to be quick. Therefore, resource issues had to be addressed including manpower and instrument/equipment availability. A high priority must be placed on the entire program by both the service provider, the client, and other involved partners (e.g. government or regulatory agencies etc.).
- Is there anything that you would do differently when employing this process in the future?
Through this practice, the team learned how to effectively and efficiently work together to achieve our goal, and those lessons learned opened another level of motivation to do better. As a case in point, and not long after the conference, WuXi Biologics, along with Tychan, announced an even shorter timeline (7-months from DNA to IND) for another infectious disease initiative. This time the program was a novel monoclonal antibody to fight Yellow Fever. We are very proud to have accomplished this goal for our partner and we are looking forward to working with our other clients to help bring better therapeutic products to patients faster.