

Key Tools and Technology Hurdles in Advancing Stem-Cell Therapies – Part I
This white paper was sponsored by: California Institute for Regenerative Medicine Alliance for Regenerative Medicine – Cell Therapy Catapult and is reprinted in Cell Culture Dish in two parts and with permission.
Authors: Elona Baum, Neil Littman, Morrie Ruffin, Stephen Ward, Kathy Aschheim
Researchers and product developers in the field of regenerative medicine face a complex set of challenges in translating stem-cell science from the laboratory through clinical trials and ultimately to patients. The California Institute for Regenerative Medicine (1) (CIRM), the Alliance for Regenerative Medicine (2) (ARM), and the Cell Therapy Catapult (3) (Catapult)convened a roundtable on June 25, 2013 to define technological challenges and identify potential solutions in three key areas: manufacturing clinical-grade cell products derived from stem cells, characterizing the cell product, and imaging cells after transplantation. This White Paper summarizes the proceedings from the roundtable and highlights key recommendations.
I. Cell-Product Manufacturing
Discussions relating to cell-product manufacturing focused on techniques and practices used in the early stages of a manufacturing process that could create significant hurdles at later stages if requisite steps are not taken to ensure a smooth transition to current good manufacturing practice (cGMP) compliance and commercial-scale production. Ensuring that a cell-manufacturing process complies with the cGMP and chemistry, manufacturing and controls (CMC) standards of regulatory agencies means that the product is made in a safe, reproducible manner, but it does not necessarily imply that the process is sufficiently robust, scalable, and cost effective to achieve commercial success. cGMP compliance is not required at the pre-clinical stage, and developing a commercially viable manufacturing process is viewed by many as not economically feasible or premature in the absence of efficacy data. Thus, it is not uncommon for early- stage processes to include non-cGMP-grade cells, xenogeneic raw materials, open flasks, labor-intensive manual culture protocols, or other problematic elements. At later stages of development, however, such practices can create significant obstacles because they may be difficult and expensive to remove. The reason is that, from the perspective of regulators, in many respects the ‘product is the process.’ Therefore, to modify the manufacturing process as research proceeds from a phase I clinical trial, with few patients and small supply demands, to phase II and III trials, with significantly more patients and significantly larger supply demands, a clinical-trial sponsor must convince regulators that the product is the same despite the changes made to the manufacturing process. This can be a very expensive and time-consuming proposition. At best, it is achieved through an in vitro comparability study; at worst, early clinical trials must be repeated. The presence of problematic elements in a manufacturing process does not necessarily preclude regulatory approval of a clinical trial,but it increases corporate risk and chips away at the ‘investability’ of a cell-therapy asset.
Commercially Viable Manufacturing Process
Even before a manufacturing process is developed, the choice of a starting cell line can result in future challenges. For example, the cell line may not have been derived in cGMP conditions, or its provenance may not have been documented in sufficient detail with respect to the donor’s health information. A phase 1 clinical trial proposed by Geron was based on one of the Wisconsin Alumni Research Foundation (WARF) human embryonic stem cell (hESC) lines, which were not derived under cGMP conditions. In this instance, Geron was allowed to conduct the clinical trial after adventitious agent testing of the master cell bank and of the final cell product. However,the use of cell lines that lack cGMP compliance or appropriate donor medical histories adds risk with respect to eventual regulatory licensure and commercialization of the cell product. Many of the groups conducting translational research with hESCs work with WARF cell lines or lines from the UK Stem Cell Bank, and some of the roundtable participants suggested that these groups consider using different cell lines that have been derived in full compliance with donor-eligibility and cGMP guidances. Others proposed that hESCs could be replaced with induced pluripotent stem cells, although these cells would be subject to the same donor-eligibility and cGMP standards as hESCs and would raise new safety concerns related to uncertainties about the reprogramming process.
Another area for caution is the use of xenogeneic raw materials, such as fetal bovine serum and matrix preparations isolated from animals. Materials of animal origin may carry pathogens, which would compromise the safety of the cell product. In addition, xenogeneic raw materials are chemically undefined and often have excessive lot-to-lot variability, undermining process control.
Although the details of the manufacturing process will be different for most cell products,the roundtable participants recommended that researchers establish a commercially viable process as early as possible and certainly before embarking on preclinical safety and toxicology studies.More specifically, considering that the most expensive parts of a manufacturing process are often the problematic elements that must be removed, it is important to identify and eliminate such elements at an early stage, in accordance with a risk/benefit analysis. While it may be tempting to defer the task of establishing a commercially viable manufacturing process because of cost considerations or a perceived need to prioritize proof-of-concept studies, this is not advisable as it could derail the development process in the future.
To construct a robust manufacturing process, one should begin by dividing the process into unit processes and then specify the inputs and outputs of each unit. Unit processes form the basis for devising standard operating procedures, in-process controls, and release criteria that define the product. Yet even when standard operating procedures are in place, minor differences in how they are executed by different technicians can lead to unacceptable variation between cell lots. Therefore, some of the roundtable participants proposed that ultimately it would be desirable to replace human technicians with automated, robotic systems. Of course, the feasibility of automating a given process would depend on a favorable cost-benefit analysis and on whether the process is understood in sufficient depth. As the cell-therapy industry matures, it will likely move from simpler forms of automation, such as liquid handling robots, to more sophisticated systems involving full-control bioreactors, in-line continuous monitoring, and enclosed harvesting. Much of this technology already exists, but more work is needed to understand how to implement it.
Scalability
The number of cells required for a stem cell–based therapy varies considerably depending on the clinical indication. Moreover, projected demand for different cell therapies ranges from relatively few patients to many millions.
It is important to recognize that scaling up cell production is not equivalent to replicating a small-scale process many times over (scaling out). Rather, it often requires new systems for culturing, expanding, and manipulating cells. If producing enough cells in adherent culture would consume surfaces the size of football fields, it may be necessary to move to suspension culture. If cell manufacture expands from one facility to several, this could entail new ways of storing, packaging or transporting cells.Scaling up cell production, if performed improperly, is incompatible with rigid process control and can create challenges in the form of belated process changes. To address this issue, the roundtable participants emphasized that researchers should evaluate potential future scale-up needs as early as possible to ensure that scalable manufacturing systems are adopted in a timely manner.
Summary of Technology Opportunities in Product Manufacturing
The roundtable participants identified a set of tools and technologies for product manufacturing that, if developed, would be broadly beneficial to the cell-therapy industry. Table 1 in Section V summarizes these key technology opportunities, which include:
- methods for expanding pluripotent and differentiated cells to large numbers (> 10¹²)in suspension culture.
- approaches for making culture conditions more hospitable to cells, through research on the biology of the cell microenvironment,or niche, including ‘smarter’ bioreactors with better in-process control and feedback loops.
- methods for enclosed volume reduction without centrifugation.
- methods for improving yield during cell isolation.
- small molecules to replace growth factors and cytokines.
- synthetic matrices to replace biological ones.
- methods to provide cells in final formulation media.
- other strategies for achieving better control over the purity and identity of the product and for understanding the effects of variations.
A Best Practices Tool Box to Address Technology Opportunities
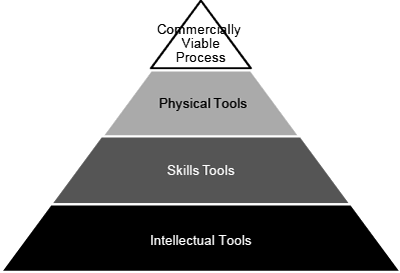
Future technology innovations in areas such as these are expected to lower manufacturing costs over time and accelerate the development process. In addition to ongoing technological progress,the roundtable participants concluded that the cell-therapy industry as a whole would benefit immensely froma concerted effort to create a publicly accessible ‘Tool Box’ that would define and disseminate practices that are likely to support successful commercialization efforts. As shown in Figure 1, the Tool Box can be thought of as containing several types of tools: the goal of a commercially viable process is achieved through the physical tools needed to produce the cell product, the skills tools needed to use the physical tools, and the intellectual tools needed to design and manage the overall program. Ideally, the tools would be validated by organizations such as the US Food and Drug Administration (FDA), CIRM, ARM, Catapult, National Institute of Articless and Technology, US Pharmacopeial Convention,and the UK Medicines and Healthcare Products Regulatory Agency.
Thus, the Tool Box would contain both physical resources (cells, media, instruments, etc.) and documentation on best practices. An example of the latter is Quality by Design, a strategy developed by the FDA to build product quality into a manufacturing process and to enable risk-based decisions throughout the development life-cycle. Rather than testing a cell product for quality at the end of the production process, Quality by Design applies statistical and risk-management tools to define ‘critical process parameters’ that determine a product’s ‘critical quality attributes.’ When this approach is used correctly, the result should be a strong understanding of how process changes affect product characteristics.
The roundtable participants highlighted six specific areas in the cell- manufacturing process where it is particularly important to ma
ke decisions with a longer-term view and that could be prioritized in developing tools to be included in the Tool Box (Figure 2 and Table 2). Although the six examples pertain to cell manufacturing, the Tool Box could also contain tools relevant to cell characterization,in vivo imaging and other aspects of cell-therapy development.
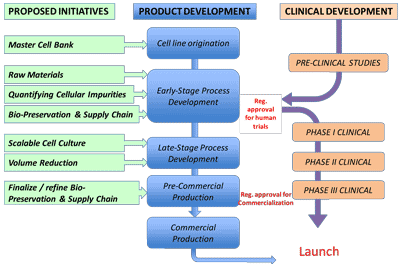
| Tool | Rationale |
|---|---|
| Master cell bank | The foundational cells must be derived in a controlled, documented, pathogen-free environment with appropriate consent and medical history documentation. |
| Raw materials | Materials contacting process intermediates or product must, where possible, be manufactured under cGMP and tested for quality. |
| Cell impurities quantification methods | Quantification of impurities is essential for demonstrating the safety of high-dose cell therapies. |
| Bio-preservation and supply chain systems |
Refrigerated or ambient storage systems would enable worldwide distribution of cell products. |
| Scalable cell culture methods | Large-scale cell therapies require multi-thousand-liter, homogeneous culture systems and, for autologous products, robust platform processes. |
| Volume reduction methods | This unit operation bridges upstream and downstream processing with product formulation, fill, finish, and distribution. |
Please click for Part II