

Upstream Manufacturing of Gene Therapy Viral Vectors

Overview
The majority of gene therapy applications in development utilize viral vectors to carry the therapeutic gene into the target cells. Cells may be genetically modified either in vivo or ex vivo. In the ex vivo applications cells are modified in culture, which also allows for cell expansion and analytical characterization prior to re-infusion of the treated cells. Typically, cells are examined pretreatment and post treatment for viability, density, expression level, etc. The ex vivo process can sometimes result in more efficient transduction.
There are several possible viral vectors systems available and the decision of which to use depends on many factors that include tissue tropism, desire for integrating or non-integrating modification, in vivo or ex vivo process, prior immune exposure, whether the target cell is replicating or non-replicating, and safety. High efficiency transduction and robust levels of transgene expression are desired outcomes in applications with viral vectors.
One of the greatest safety concerns with viral vectors is generation of wild type infectious virus from vector components. For this reason, viral vectors are typically manufactured from 2, 3 or even several separate expressible units to significantly reduce the possibility of forming a wild type particle via recombination. The expressible units are typically separate plasmids introduced to producer cells either by transfection or through “helper” transducing viruses. The expression units are usually further engineered with mutations to disable the function of the wild type virus should they be formed by a recombination event. For example, newer HIV lentiviral vectors delete genes for virulence factors tat, vpr, vpu, nef and/or have gag and pol on separate plasmids from rev and env1 and/or contain other mutations such as deletions in the 3’ LTR.2
The overall goal in viral vectors design is to efficiently package the therapeutic gene or nucleotides into infectious viral particles and avoid the generation of wild type particles or empty particles. Thus, many vector systems are manufactured by transient co-transfection of multiple plasmids for either safety, for convenience, or by necessity in order to avoid toxicity of a vector component in producer cells (Figure 1).
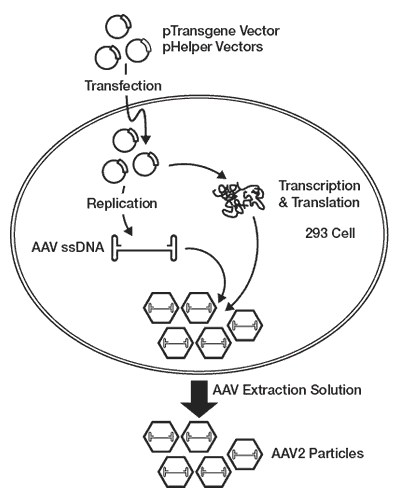
AAV vector components and therapeutic gene are transfected, usually as separate expression cassettes from different plasmids. Expression cassettes are expressed within the cell resulting in viral proteins and a genomic ssDNA containing the expression cassette for the therapeutic gene(s). In the AAV system, viral particles containing the transgene assemble in the cytoplasm. Particles are released to the media via cellular lysis prior to further purification and characterization.
Major Viral Vectors in Use
Historically, there has been more focus on retroviral and adenoviral vectors, which have now generated a 30-year history.3 However, because of enhanced safety and improved target tissue expression profiles, vectors derived from adeno-associated virus (AAV) and lentiviral vectors have advanced in development. Herpes simplex virus and pox/vaccinia vectors also show promise for their effectiveness as oncolytic vaccines.
AAV is the most commonly used vector for in vivo genetic modification, and different serotypes (naturally occurring or recombinant) can be used to target different tissues within the body.4 AAV is not known to be pathogenic and is thought to be well tolerated, resulting in lower inflammatory response.5,6 Moreover, unlike other viral vectors such as lentivirus that integrate into the host cell genome, AAV is thought to primarily remain episomal.3,7
One downside to AAV is its low transgene packaging capacity relative to other viruses5 but this is not currently a major problem with the diseases being targeted or investigators have designed mini-genes (that fit within AAV) for the disease targets. AAV also has a propensity to package any and all DNA in the vicinity including host cell DNA and plasmid backbone DNA.8, 9
For ex vivo genetic modification, lentiviral vectors are now the most commonly used. Lentiviral vectors have advantage over classical gamma retroviral vectors because they can transduce both dividing and non-dividing cells. Lentiviral transduction of slowly dividing CD34+ HSCs has been applied to several genetic diseases, including β-thalassemia10, X-linked adrenoleukodystrophy11, and metachromatic leukodystrophy.12 There are also fewer challenges associated with insertional oncogenesis compared to gamma retroviral vectors (MuLV) like those that hindered early clinical progress with gene therapies retroviral vectors.1,13
The table below summarizes some physical properties of the various viruses that are used for gene therapy:
| Parameter | Retrovirus | Lentivirus | AAV | Adenovirus |
|---|---|---|---|---|
| Coat | Enveloped | Enveloped | Non-enveloped | Non-enveloped |
| Packaging capacity (Kb) | 8 | 8 | ~4.5 | 7.5 |
| Tropism/infection | Dividing cells | Broad | Broad excluding hematopoietic stem cells | Broad |
| Inflammatory potential | Reduced | Reduced | Reduced | High |
| Host genome interaction | Integrating | Integrating | Integrating/ non-integrating | Non-integrating |
| Transgene expression | Long lasting | Long lasting | Potentially long-lasting | Transient or long-lasting depending on immunogenicity |
Overview of Viral Vector Manufacturing
The manufacture of viral vectors may require several manufacturing phases or platforms. Initially, the materials needed to manufacture the therapeutic viral vector must be generated. These include plasmids encoding helper-virus functions and the therapeutic gene, cell lines used to manufacture the vector, and other materials (See Figure 2, plasmid manufacturing). In some cases, helper transducing viruses may be substituted for plasmids. Alternatively, stable producer cell lines may be created in order to reduce or eliminate the transfection and/or transduction steps and simplify the production process.
The next step in vector manufacturing involves the generation of infectious viral vector (Figure 2, viral manufacturing). Cells are transfected with plasmids to generate the viral vector, which is harvested. AAV viral particles accumulate in the cytoplasm and the media, so total yield can be enhanced by lysing the cells. Harvested vector is then concentrated, purified, titrated, characterized, and stored for later ex vivo or in vivo used.
In ex vivo transduction (Figure 2, cell processing), target cells are collected and modified by the viral vector. Following modification, the cells are harvested, characterized, and formulated prior to transplantation. In some processes, transduced cells may be expanded in cell culture prior to the re-infusion into the patients.
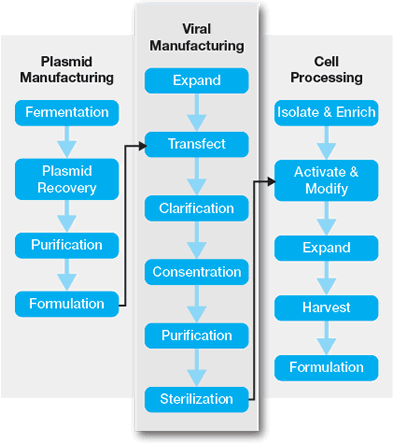
Generation of Viral Vectors by Transfection, Infection, or by Stable Producer Cells
In general, there are two modes of vector production: transient production systems or producer cell lines, which may be mammalian or insect cells in the case of baculovirus vectors. Transient production systems involve either transfection of one or several plasmids encoding the helper virus functions or alternatively transient systems may use other viruses to provide helper function.14 At the present time, many vectors are currently generated by transient transfection either due to convenience, expense, or because of the lack of a stable producer cell line.
Manufacturing viral vectors by transfection offers advantages and disadvantages. It is flexible and efficient, does not require time-consuming development of stable cell lines, and allows for successful vector generation should any viral component produce cellular toxicity upon expression.15 However, transfection-based methods have disadvantages due to the requirement for costly GMP grade plasmids for each transfection lot and the possible requirement of additional purification steps to remove plasmid and cellular DNA. Moreover, transfection of suspension cells at large scale can prove difficult and can result in relatively low yields.
Among the transfection methods, polyethylenimine (PEI) transfection has advantages over calcium phosphate since it is less toxic and may eliminate the need for a media exchange. PEI transfection is also less dependent on pH, the presence of serum, and is effective for both adherent and suspension cultures.16 Drawbacks are the amount of costly plasmid required for large suspension culture and the absence of an analytical method to quantify PEI in the purified vector preparation.17 Lipid-based transfection reagents are also effective; however, expense may make them impractical for use at larger scales.18 Non-chemical methods such as electroporation show promise, however, the need to concentrate cells can also make its use impractical at large scales.17 Transfection-based methods, while fast and efficient, can pose significant challenges, particularly at larger scale manufacture.
Use of Baculoviral Vectors for Therapy or to Produce Other Viral Vectors
Low toxicity and inability of baculoviruses to replicate in mammalian cells make them potential candidates for therapeutic gene delivery.19 Baculovirus-mediated gene delivery into dividing and non-dividing mammalian cells has demonstrated therapeutic efficacy in both ex vivo and in vivo gene therapy studies.20, 21, 22
A suitable expression cassette and/or a pseudotyped envelope such as VSV-G can be used to transduce various cell types.23, 24 Baculoviral vectors can mediate high-level transient expression of transgenes in many stem cells25can be modified to permit stable transgene expression.26 The high-level of transgene expression from baculoviral vectors is well suited for cancer gene therapy22,27 and the lack of pre-existing antibodies to baculovirus in humans is an advantage for in vivo therapy.
Baculoviral vectors have advantages and disadvantages. Baculoviral vectors offer an attractive alternative to transfection for vector generation and have been used in the production of other vectors such as AAV from insect cells28 lentiviral vectors from suspension 293T cells.29 Baculovirus and other infection-based systems for the generation of vector are typically easier to work with at large scale than transfection but require additional manufacturing steps in order to produce the baculoviral vector. There are some drawbacks to the use of baculoviral systems. Some cell culture media formulations can result in reduced transduction efficiency.20 Another challenge with this approach is that health authorities are increasingly interested in ensuring minimal levels of residual baculovirus (infectious or not) remain in the final product.
Overall, there is much promise for the use of baculoviral vectors in the future.
Stable Producer Cell Lines
Cellular toxicity from the expression of a required vector component has complicated the establishment of stable producer cell lines for many vector systems. Some stable producer lines utilize inducible promoters to minimize toxicity, and most stable MuLV retroviral cell lines are inducible.14 One example of cellular toxicity produced from a vector component is the pseudotyped retroviral envelope VSV-G.1
Increasingly, the field is thinking that stable cell lines with inducible systems to express the viral component is the way to go. Toxicity is one aspect. Another difficulty is the prospect of making infectious virus without any nucleotides packaged. That effectively becomes an impurity that must be cleared via the purification process.
Similarity, toxicity from lentiviral components (pol, rev) has thwarted the development of stable lentiviral producer cell lines. Only recently are methods being established for generating clinical-grade cell lines that continuously produce lentiviral vector.30
The development of stable cell lines is expected to significantly improve the current challenges associated with producing viral vector at scales needed for therapies with large therapeutic demand or those requiring high titers of vector. The use of a stable cell lines simplifies manufacturing steps, lessens regulatory oversight, and lowers cost. This, in turn, could significantly enable the development of new vector-based gene therapies.
Choice of Producer Cell Line
Many factors influence the choice of cell line for vector generation. A key factor is production titer. Increased production titers of therapeutic vector can greatly reduce both production scale and lower costs. For vectors produced by transfection, transfection efficiency is a major factor. HEK 293 and HT1080 cells can be transfected at efficiency of near 90% or they also produce high titers of vector.14 Among the many HEK lines, HEK 293T cells reportedly produces increased titer because of the presence of SV40 T-Ag.1 Recently, “Fast” HEK 293FT cells, which reportedly have shorter doubling times and greater production are showing promise.31 Many companies are actively expanding their repertoire of available cell lines by exploring unconventional sources such as engineered human cell lines with minimal prior viral infection.
Scaling of Adherent Cells for Manufacturing Vector
The majority of the cell lines used to generate viral vectors are naturally adherent, with the exception of some tumor cell or blood lines which can grow in suspension.14 Adherent cells are limited by the surface area of the device. Hence, production scaling is accomplished either by increasing the number of identical culture systems units (flasks, roller bottle, cubes, HYPERStacks®) (scale out) or using successively larger devices (scale up).32
Much work to date with viral vector manufacturing has used adherent based processes generally in multilayer cell factories (CF) and HYPERFlasks/Stacks (HY) (Corning). Cell factories have been used for the production of preclinical and clinical vector batches of γ-retroviral, lentiviral33 or AAV vectors.34 CFs can provide up to 2.5 m2 a significant increase over a roller bottle with area up to 0.17 m2.
Cell factories, HY, and roller bottles are unit production systems. Any significant increase in production capacity requires additional culture units (scale out).35 Roller bottle expansion can be assisted by automation, which is commonly used in the vaccine industry. Cell factories are more difficult to scale and limitations in gas exchange have been shown to decrease viral titer for AAV production.36 However, some cell factory systems are available in semi-closed loop providing some advantage.37
Newer, HYPERFlasks® and HyperStacks® have a membrane for gas exchange which has been shown to increase lentiviral production.38 These are more flexible than cell factories in media/surface area volumes and they offer areas of up to 1.8 m2/unit. However, these also require the addition of more units for large scale production. The Corning CellCube® system can scale up to 34 m2. However, the CellCube® system is only partly single-use.14
The use of bioreactors enable scale up options and they also have additional advantages in that it is easier to monitor and control processes, reduced record keeping, fewer unit operations, lower contamination risks, and lower operation costs.14,39,40 The improved control of culture condition in bioreactors may also result in improved productivity. Furthermore, many bioreactor systems can also be used in a perfusion mode which is an attractive option for retroviral and lentiviral production.14
Hollow fiber bioreactors (e.g., the Quantum® bioreactor, Terumo BCT)41 and fixed-bed bioreactors look promising for attachment-dependent cells as they offer increased scale42 and sometimes scalability options within a unit. For example, the iCELLis® fixed-bed bioreactor (Pall) has two models. The iCELLis® Nano system is a smaller bench top unit and the larger 500 model is a stand-alone unit (Figure 3).
The fixed-bed consists of polyester (PET) macrocarriers (13.9 cm2 each) fixed inside a housing where media flows from the bottom to the top. The height of the bed can vary as can the compaction density of carrier which enables a wide range of scalability options up to 500 m2 (Table 2). The maximum area of 500 m2 offers greatly increased scale which is equivalent to roughly 5900 roller bottles, 780 10-layer stacks, or 280 HYPERStacks (Table 2). In addition, the iCELLis® bioreactor enables high cell density growth of 108 cells/mL carrier and produces similar cell-specific titers compared to adherent culture.43
| Bed Size | Surface Area/m2 | Equivalent Units | ||||
|---|---|---|---|---|---|---|
| lBioreactor | Bed Diameter (cm) |
Bed Height (cm) |
Carrier Compaction Low-High |
850-cm2 Roller Bottles | 10-Layer CellStacks | HYPERStack 36 |
| iCELLis Nano | 11 | 2 | 0.53 – 0.8 | 6.2-9.4 | 0.04-0.08 | 0.3-0.4 |
| iCELLis Nano | 11 | 4 | 1.06 – 1.6 | 12.4-18.8 | 0.2-0.5 | 0.6-0.9 |
| iCELLis Nano | 11 | 10 | 2.65 – 4.0 | 31.8-47.1 | 10-25 | 1.5-2.2 |
| iCELLis 500/100 | 86 | 2 | 66 – 100 | 776-1116 | 104-157 | 37-56 |
| iCELLis 500/200 | 86 | 4 | 133 – 200 | 1565-2353 | 209-314 | 74-111 |
| iCELLis 500/500 | 86 | 10 | 333 – 500 | 3918-5882 | 524-786 | 185-278 |
While hollow fiber and fixed-bed bioreactor systems offer significant increases in scale, the scale is currently limited. Another option for adherent cells is the use of microcarriers which can provides a large surface area for cell attachment in rocking bioreactors or stirred tank bioreactors. Rocking bioreactors with microcarrier or single cell suspension culture are an option to produce smaller quantities of vector for research or clinical studies, as their currently capacity is limited 500L at low cell density. Rocking bioreactors are disposable and can also be used to provide cell seed for larger bioreactors.
Virus production in the vaccine industry has been successfully scaled up to 2000+L using microcarriers in stirred tank bioreactors.44 A single 2000L bioreactor operating at 10 cm2/mL microcarrier density offers 2,000 m2 of surface area. Drawbacks to microcarriers are developmental considerations of optimizing cell attachment, growth, and viability. Moreover, the ability to transfect/transduce cells growing on a microcarrier needs to be understood early in development.
Stirred-tank bioreactors are by far the most prevalent bioreactor used for the commercial manufacturing of mAbs and recombinant proteins. As a result, the technology is very well characterized and both industry and regulatory authorities are very familiar with their operation. They are the most efficient means of scaling up to large volumes. While these bioreactors can be used to grow adherent cells with the use of microcarriers, they are best adapted to the growth of suspension cells. Stirred tank bioreactors come in a variety of size and configuration options. For example, the Allegro™ STR (Pall) is a single-use bioreactor that is available in 50, 200, 1000 and 2000 L scales (Figure 4). In addition, these bioreactors have a compact footprint due to their atypical cubical design.

Scaling of Suspension Cells for Manufacturing Vector
Single-cell suspension culture has significant advantage over adherent cells in that they can easily be scaled out from spinner, to laboratory scale bioreactor, to production bioreactor without cell detachment. Several 293 cell lines used for vector production (293T, 293FT) are prone to adaptation to suspension culture in chemically defined media45 and suspension 293 cell lines preadapted to commercial media are available on the market. The use of animal-free media is a desirable feature for manufacturing due to the decreased risk of adventitious agents and the reduced purification burden. However, adapting custom cells to suspension culture can be time consuming and prolong process development time.
Currently, there are challenges associated with transfection of cells at scales >200L which is a significant limiting factor to widespread adoption of suspension culture for processes dependent on transfection.17 More recently, published reports have proven that high titers of lentiviral vector can be achieved using a suspension adapted helper cell line in small scale bioreactors.46 Although promising, methods using suspension cells at larger scale have not yet been widely adopted for routine vector production.
Cost Considerations
There are several factors that can influence the cost of viral based gene therapy manufacturing. These include production titer, the ability to upscale production processes, regulatory burden, and other factors.
Production titer is a key factor as higher titer results in a smaller, more cost-effective production process. High titer production of vector can lower reagent demand, labor, and facility requirement. One possible route to increase titer is by control of cell environment and production conditions. In this regard, controlled bioreactor vessels can present an advantage over flasks, roller bottles, or CF’s in productivity.
In addition, use of bioreactors can simplify scale up of a process as previously described. From a capex, point of view, scaling up multilayer stacks, requires considerable space. Since clean room space is expensive, this can be a very significant cost. In contrast, implementation of a bioreactor system can be done in the same space required for a modestly size multilayer stack system.
Implementation of a bioreactor system can decrease labor and consumables costs 3-fold and there is considerably less plastic waste generated with a bioreactor system resulting in reduced disposal costs. Most bioreactor systems come with some degree of automation which manifests itself as a 3-fold decrease in labor compared to more manual systems like multilayer stacks. Overall a >50% reduction in costs can be achieved by implementing a bioreactor system compared to a multilayer stack of similar scale.47 Scaling out a process that uses adherent cells in multilayer CF requires many connections and incubators, so contamination risk increases along with capex costs. Finally, disposable systems like the iCELLis® bioreactor, or other single use systems can provide savings as cleaning, sterilization as well as extensive validations are reduced or eliminated.
Suspension bioreactors also have lower risk due to the reduction of production units, especially compared to a scale-out, multi-flask configuration. With very small clinical trial material requirements, adherent cell technology may suffice with some cost savings on capex.
Process Design and Regulatory Burden as a Cost Determinant
Production design can have a large effect on cost. While quick, easy, and flexible, vector production by transfection poses both increased cost and challenges as scale increases. The use of helper viruses (for direct infection) rather than plasmids may reduce transfection needs and provide a path for scaling up production. In this regard, the use of a stable producer cell line would provide the greatest benefit through the reduction of manufacturing steps. The use of a stable producer cell line over transfection methods may also facilitate process consistency, which is an attribute for GMP vector production.48 However, the use of inducible cell lines may require additional steps to remove inducing agents.
Since all components of vector production intended for therapy require GMP manufacture, optimization or elimination of costly GMP steps, such as the manufacture of plasmid, provides large benefit.49 Design optimization of the overall process in light of regulatory burden for purity and safely could significantly lower production costs.50 These include the presence of potentially toxic or immunogenic impurities, contaminants such as HCP proteins and DNA, plasmid DNA, reagents from transfection, potential transduction inhibitors, induction agents, antibiotics, and others.51 Other areas include screening for producer cells and vector batches for replication-competent viruses.48Consideration of vector sterilization may also reduce costs given that large viruses, such as HSV, may not be filter sterilizable, thus requiring aseptic processing, and validation that goes with that. As some gene therapy products are relatively new for regulatory authorities, it is highly likely that regulatory guidance will increase as more products are taken to market.50
Process Development Timelines
Adherent based bioreactor systems such as the iCELLis® have advantages over suspension systems in terms of process development timelines. Adaptation of a process from cell factories to adherent based bioreactors does take some effort but timelines can be relatively short. By comparison, converting an adherent system to suspension can be a very lengthy process (more than 1 year). Other possible complications are variation in product quality that would requiring extensive comparability testing because of biological differences between adherent and suspension systems.
Transfection based approaches are quick and flexible. However, they are expensive and more difficult to upscale. Transfection approaches may have benefit in that vector can be produced quickly to enter clinical evaluation. However, this approach may provide technical and cost difficulties for greater upscaling for vectors destined for wide therapeutic distribution.
Adapting an adherent system to a fixed-bed bioreactor does take some time but this is generally much shorter than adapting adherent systems to suspension, which can be very difficult and may result in decreased productivity.
Analytical Considerations
The final vector product intended for gene therapy and vaccine applications needs to be well characterized and of proper quantity, purity and potency. This can be achieved only if adequate characterization methods are in place and the viral production process is well established, understood and reproducible. Sample control and product characterization are not only important for the final product, but also during different steps within the production process. In depth knowledge of both up- and downstream processing is crucial since the upstream production greatly affects the downstream process. Sometimes even slight changes in the upstream process can result in a less efficient downstream process.
The well-established framework for traditional biologics (i.e. recombinant proteins, enzyme replacement therapies, monoclonal antibodies) is increasingly being applied to the field of gene therapies. As such, more investment in real-time, high-throughput technologies to characterizer both process and product intermediates is needed. The days of process = product are over and a new paradigm is needed to bring the potential of gene therapies to patients in need.
Impact on Downstream Purification
Many vectors are secreted into the media. In this case, virus can be collected from the media which will substantially reduce downstream burden. In some cases, a portion of the virus remains in the cells and so cell lysis with either detergent or physical means may be critical to improve yield. There are a wide variety of processes that can be used for purification, including clarification by filtration or centrifugation; chromatography steps for affinity, ion exchange, sizing, multi-modal and other resins; concentration steps with TFF or ultracentrifugation; and enzymatic treatments to reduce nucleic acids. When the cells must be lysed to release the vector products, multiple steps may be needed to optimize the purification.
The choice of upstream production process can affect downstream purification burden. For example, shear forces on adhered cells on microcarrier systems or other systems with fluid motion can increase cell debris. Another example is that a fixed-bed bioreactor can sometimes offer some benefit some impurities can be trapped into the bed.
Traditional purification steps have relied on ultracentrifugation, flocculation, and other difficult-to-scale technologies. Recent advances have enabled the same level of impurity removal to be obtained using highly scale-able chromatography and tangential flow filtration.
Room for Improvement
There are several areas of improvements for viral vectors for gene therapy. As mentioned earlier, transfection can be both problematic and expensive at larger scales required for wide clinical demand. As such, advancement in the use of stable producer lines, particularly in the HIV lentiviral system is needed. The cost of vector and vector-derived therapies continues to present a challenge towards advancement. Methods need to be developed which result in lower costs.
The development of single-cell stable producer cell lines may further increase production scale, and in particular if the cell can be adapted to growth in suspension, by enabling the use of large tank bioreactors. These, in turn, can be used in closed systems with less risk of contamination. Moreover, the integration of serum-free media will lessen purification burden and animal-free media will improve regulatory safety profiles.
Current methods for lysing cells (example AAV) are also crude and there is room for improvement. Indeed, Triton™-X, which is traditionally one of the most widely used lysing agents, has restrictions on use in the EU.52 In addition, there are often shortages of commonly used items, such as tissue culture plastic ware used for expanding adherent cells due to the very rapid increase in the number of gene therapy projects.
As more serotypes of certain vectors are being designed for more tissue specific uses there is an increased need for more generic and specific affinity reagents and new techniques for vector purification.
Despite the ongoing need for improvements in therapeutic vector production, there is much optimism for the future of viral based gene therapy. In the future, vector based gene therapy may not only offer gene replacement, but could advance to offer gene editing functions as well.
This article is the third in a series that was published in our eBook, “Key Considerations for Gene Therapy Manufacturing for Commercialization“. You can download all the articles in the series, by downloading the eBook below.
Footnotes
-
1. Milone, M.C. and U. O’Doherty, Clinical use of lentiviral vectors. Leukemia, 2018. 32(7): p. 1529-1541.
-
2. Dull, T., et al., A third-generation lentivirus vector with a conditional packaging system. J Virol, 1998. 72(11): p. 8463-7
-
3. Hastie, E. and R.J. Samulski, Adeno-associated virus at 50: a golden anniversary of discovery, research, and gene therapy success--a personal perspective. Hum Gene Ther, 2015. 26(5): p. 257-65.
-
4. Balakrishnan, B. and G.R. Jayandharan, Basic biology of adeno-associated virus (AAV) vectors used in gene therapy. Curr Gene Ther, 2014. 14(2): p. 86-100.
-
5. Colella, P., G. Ronzitti, and F. Mingozzi, Emerging issues in AAV-mediated in vivo gene therapy. Mol Ther Methods Clin Dev, 2018. 8: p. 87-104.
-
6. Merten, O.W., AAV vector production: state of the art developments and remaining challenges. Cell GeneTherapy Insights, 2016. 2(5): p. 521-551.
-
7. Srivastava, A., Advances and challenges in the use of recombinant adeno-associated virus vectors for human gene therapy. Cell GeneTherapy Insights, 2016. 2(5): p. 553-575.
-
8. Chadeuf, G., et al., Evidence for encapsidation of prokaryotic sequences during recombinant adeno-associated virus production and their in vivo persistence after vector delivery. Mol Ther, 2005. 12(4): p. 744-53.
-
9. Schnodt, M., et al., DNA minicircle technology improves purity of adeno-associated viral vector preparations. Mol Ther Nucleic Acids, 2016. 5: p. e355.
-
10. Cavazzana-Calvo, M., et al., Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature, 2010. 467(7313): p. 318-22.
-
11. Cartier, N., et al., Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science, 2009. 326(5954): p. 818-23.
-
12. Biffi, A., et al., Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science, 2013. 341(6148): p. 1233158.
-
13. Hematti, P., et al., Distinct genomic integration of MLV and SIV vectors in primate hematopoietic stem and progenitor cells. PLoS Biol, 2004. 2(12): p. e423.
-
14. Merten, O.W., et al., Manufacturing of viral vectors for gene therapy: part I. Upstream processing. Pharm. Bioprocess., 2014. 2(2): p. 182-203.
-
15. Krieg, A.M., Direct immunologic activities of CpG DNA and implications for gene therapy. J Gene Med, 1999. 1(1): p. 56-63.
-
16. Segura, M.M., et al., New protocol for lentiviral vector mass production. Methods Mol Biol, 2010. 614: p. 39-52.
-
17. Merten, O.W., M. Hebben, and C. Bovolenta, Production of lentiviral vectors. Mol Ther Methods Clin Dev, 2016. 3: p. 16017
-
18. Cribbs, A.P., et al., Simplified production and concentration of lentiviral vectors to achieve high transduction in primary human T cells. BMC Biotechnol, 2013. 13: p. 98.
-
19. Kost, T.A., J.P. Condreay, and D.L. Jarvis, Baculovirus as versatile vectors for protein expression in insect and mammalian cells. Nat Biotechnol, 2005. 23(5): p. 567-75.
-
20. Mahonen, A.J., et al., Culture medium induced vimentin reorganization associates with enhanced baculovirus-mediated gene delivery. J Biotechnol, 2010. 145(2): p. 111-9.
-
21. Madhan, S., M. Prabakaran, and J. Kwang, Baculovirus as vaccine vectors. Curr Gene Ther, 2010. 10(3): p. 201-13.
-
22. Wang, S. and G. Balasundaram, Potential cancer gene therapy by baculoviral transduction. Curr Gene Ther, 2010. 10(3): p. 214-25.
-
23. Volkman, L.E. and P.A. Goldsmith, In vitro survey of Autographa californica nuclear polyhedrosis virus interaction with nontarget vertebrate host cells. Appl Environ Microbiol, 1983. 45(3): p. 1085-93.
-
24. Tani, H., et al., In vitro and in vivo in vivo in vivo in vivo gene delivery by recombinant baculoviruses. J Virol, 2003. 77(18): p. 9799-808.
-
25. Ho, Y.C., et al., Transgene expression and differentiation of baculovirus-transduced human mesenchymal stem cells. J Gene Med, 2005. 7(7): p. 860-8.
-
26. Ramachandra, C.J., et al., Efficient recombinase-mediated cassette exchange at the AAVS1 locus in human embryonic stem cells using baculoviral vectors. Nucleic Acids Res, 2011. 39(16): p. e107.
-
27. Luo, W.Y., et al., Development of the hybrid Sleeping Beauty: baculovirus vector for sustained gene expression and cancer therapy. Gene Ther, 2012. 19(8): p. 844-51.
-
28. Urabe, M., C. Ding, and R.M. Kotin, Insect cells as a factory to produce adeno-associated virus type 2 vectors. Hum Gene Ther, 2002. 13(16): p. 1935-43.
-
29. Lesch, H.P., et al., Production and purification of lentiviral vectors generated in 293T suspension cells with baculoviral vectors. Gene Ther, 2011. 18(6): p. 531-8.
-
30. Sanber, K.S., et al., Construction of stable packaging cell lines for clinical lentiviral vector production. Sci Rep, 2015. 5: p. 9021.
-
31. Fisher, T. 2018; Available from: Thermofisher.com/order/catalog/product/R70007.
-
32. Segura, M.M., et al., New developments in lentiviral vector design, production and purification. Expert Opin Biol Ther, 2013. 13(7): p. 987-1011.
-
33. Merten, O.W., et al., Large-scale manufacture and characterization of a lentiviral vector produced for clinical ex vivo gene therapy application. Hum Gene Ther, 2011. 22(3): p. 343-56.
-
34. Allay, J.A., et al., Good manufacturing practice production of self-complementary serotype 8 adeno-associated viral vector for a hemophilia B clinical trial. Hum Gene Ther, 2011. 22(5): p. 595-604.
-
35. Spier, R., Monolayer growth systems: heterogeneous unit processes., in Animal Cell Biotechnology,, R. Spier and J. Griffiths, Editors. 1985, Academic Press: London.
-
36. Okada, T., et al., Large-scale production of recombinant viruses by use of a large culture vessel with active gassing. Hum Gene Ther, 2005. 16(10): p. 1212-8.
-
37. Pakos, V. and A. Johansson, Large scale production of human fibroblast interferon in multitray battery systems. Dev Biol Stand, 1985. 60: p. 317-20.
-
38. Kutner, R.H., et al., Simplified production and concentration of HIV-1-based lentiviral vectors using HYPERFlask vessels and anion exchange membrane chromatography. BMC Biotechnol, 2009. 9: p. 10.
-
39. Ghani, K., et al., Generation of a high-titer packaging cell line for the production of retroviral vectors in suspension and serum-free media. Gene Ther, 2007. 14(24): p. 1705
-
40. Merten, O.W., State-of-the-art of the production of retroviral vectors. J Gene Med, 2004. 6 Suppl 1: p. S105-24.
-
41. Sheu, J., et al., Large-scale production of lentiviral vector in a closed system hollow fiber bioreactor. Mol Ther Methods Clin Dev, 2015. 2: p. 15020.
-
42. Lennaertz, A., et al., Viral vector production in the integrity® iCELLis® single-use fixed-bed bioreactor, from bench-scale to industrial scale. BMC Proc., 2013. 7(S6): p. 59.
-
43. Lennaertz, A., et al., Viral vector production in the integrity® iCELLis® single-use fixed-bed bioreactor, from bench-scale to industrial scale. BMC Proc., 2013. 7(Suppl 6): p. 59.
-
44. Barrett, P.N., et al., Vero cell platform in vaccine production: moving towards cell culture-based viral vaccines. Expert Rev Vaccines, 2009. 8(5): p. 607-18.
-
45. Dormond, E., et al., An efficient and scalable process for helper-dependent adenoviral vector production using polyethylenimine-adenofection. Biotechnol Bioeng, 2009. 102(3): p. 800-10.
-
46. Manceur, A.P., et al., Scalable lentiviral vector production using stable HEK293SF producer cell lines. Hum Gene Ther Methods, 2017. 28(6): p. 330-339.
-
47. Science, P.L., Cost modeling of upstream production process of lentiviral vectors in HEK-293 cells comparing multi-tray 10 stacks and fixed-bed bioreactor. Poster ESACT 2007, 2017.
-
48. McCarron, A., M. Donnelley, and D. Parsons, Scale-up of lentiviral vectors for gene therapy: advances and challenges. Cell GeneTherapy Insights, 2017. 3(9): p. 719-29.
-
49. Ausubel, L.J., et al., Production of CGMP-Grade Lentiviral Vectors. Bioprocess Int, 2012. 10(2): p. 32-43.
-
50. White, M., et al., A guide to approaching regulatory considerations for lentiviral-mediated gene therapies. Hum Gene Ther Methods, 2017. 28(4): p. 163-176.
-
51. Rodrigues, T., et al., Purification of retroviral vectors for clinical application: biological implications and technological challenges. J Biotechnol, 2007. 127(3): p. 520-41.
-
52. CRS. CRS-Chemical inspection and regulation service -REACH Restriction. 2018; Available from: cirs-reach.com/REACH/REACH_Restriction.html.