

Viral Vector Characterization: A Look at Analytical Tools

Introduction
As cell and gene therapy field continues to push products through the clinical landscape, the need to develop appropriate analytical tools has become increasingly evident. Because of their intended use in patients, gene therapy products must meet rigorous safety guidelines highlighting the importance of well-characterized analytics. The FDA introduced the Process Analytical Technology (PAT) initiative in 2004 for biopharmaceutical manufacturing, to emphasize the importance of in-depth monitoring/testing throughout the production process that goes beyond the traditional approach of process validation and extensive end product testing. The initiative has encouraged pharmaceutical companies to investigate the use of new analytical technologies to perform timely measurements of the critical quality attributes allowing for better understanding and control during the manufacturing process.1
For cell and gene therapy, one critical aspect of the supply chain is the vector used to introduce the gene of interest. Most commonly, viral vectors, derived from lentivirus (LV), adenovirus (AV) or adeno-associated virus (AVV) are used for these therapeutics. One of the challenges of characterization and quality control testing of viral vectors is their high degree of complexity. Even recombinant Adenovirus associated virus (rAAV), the smallest and least complex type of recombinant viral vectors, has a structure more complex than the most complex recombinant proteins. Key to the development of large-scale, optimized production, harvesting and purification strategies to meet the demand for viral vectors, is having accurate and reproducible analytical tools in place to monitor quality attributes ensuring a safe, high quality, consistent and efficacious product.
This article aims to provide an overview of analytical methods, with input from industry experts, used to characterize viral vectors to ensure their safety, potency, and purity to keep pace with the demands from the fast-growing field of cell and gene therapy.
A Brief Overview of Viral Vector Manufacturing
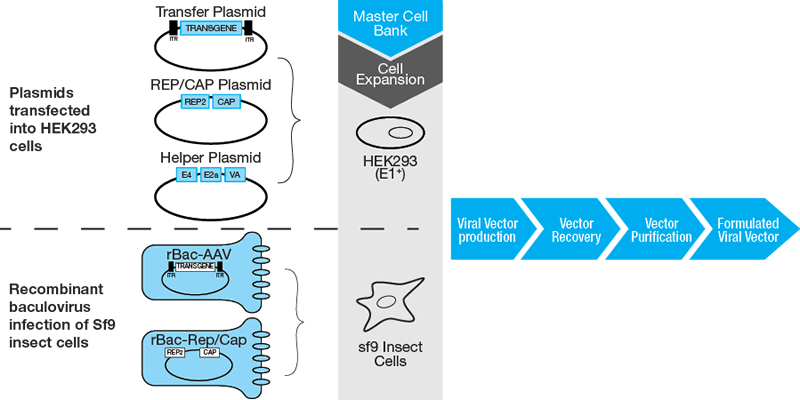
As the demand for gene therapy vectors in clinical therapeutics grows, robust, reproducible large-scale manufacturing platforms are needed (Figure 1). This may necessitate changes to:
- Components (i.e. changes in raw materials to improve traceability or compliance)
- Cell lines (i.e. adaptation to suspension cultures, use of microcarriers)
- Equipment (to increase scale of production)
- Manufacturing processes (i.e. optimize methodologies or to integrate automation)
- Manufacturing facilities
Changes in these parameters may require comparability studies to ensure that the safety, identity, purity and efficacy of the product remains unaffected. Scaling up from a research-scale protocol requires process development time to ensure all the critical quality attributes (CQAs) of the therapeutic product are maintained. The CQAs that are important to monitor during viral vector manufacturing include viral potency, identity, quantity, process residuals (i.e. Triton and deoxiribonuclease), aggregation, empty capsids, protein content and product safety. There are different characterization assays available and their applicability depends on the type of virus purified as well as the expression system used.
It is important to point out the upstream considerations prior to manufacturing that should be addressed, such as sourcing suitable raw materials (i.e. plasmids, vectors, reagents) and utilizing qualified cell and viral banks. The regulatory bodies mandate that that all components used during the manufacture are qualified and suitable for the intended purpose. After manufacturing, QC testing is performed to ensure the viral vector meets preset specifications for safety, identity, purity, potency, and stability and safety parameters. This information is combined into a release document (a Certificate of Analysis that lists the tests, specifications, and results) which is submitted to regulatory agencies, such as the FDA, for review.
During process development/optimization, the FDA mandates that overall product characterization under cGMP (current Good Manufacturing Practice) follows SISPQ (Safety, Identity, Strength/Potency, Purity and Quality):
- Safety: The product does not elicit unexpected side effects when used appropriately in the patient.
- Identity: The product is exactly what is described on the label.
- Strength/Potency: The product delivers the correct dosage over the shelf-life of the product.
- Purity: The product is free from physical, biological and chemical contamination.
- Quality: The product is manufactured using established quality systems to ensure product consistency and quality specifications are met.
There are some common quantification methods used by manufacturers to monitor key attributes to ensure specifications are met during the process development phase and beyond. These specifications should be established early on but should also be appropriate to the stage of product development, because release criteria will be refined and tightened as the product moves through clinical phases towards licensing. It is important to stress the need for high quality, properly characterized standards and controls for assay development, assay qualification, and product testing. Implementation of the appropriate standards for comparability studies that conform to cGMP is critical to overall success.
Analytical Assays
Because of the complexity of biomanufacturing of viral vectors, there can variability in the manufacturing that necessitates in-process product characterization to ensure lot-to-lot consistency. Gene therapy products must meet rigorous safety guidelines highlighting the importance of well-characterized analytics. Table 1 summarizes the assays that will be discussed in more detail in this section.
Identity and h3otency
Identity
Tests can be run to ensure that the identity of the viral vector preparation. Assays such as SDS-PAGE, mass spectrometry, immunoblotting and ELISA can be used to look at viral protein expression. The proper number, molecular weight, and stoichiometry of the viral proteins can be used to positively identify the vector. Conversely, the vector genome can be evaluated using PCR or high throughput NGS (next generation genome sequencing) to ensure positive identity.
Physical Titer
Physical titer calculates the total number of alive and dead viral particles present and is expressed as the number of viral particles per mL (VP/mL), or for AAV as genome copies per mL (GC/mL). Clinical dosing of rAAV therapeutics is usually based on physical titer, thus having accurate quality control methods that determine the amount of vector being administered is important.2 There are a variety of methods used to determine the physical titer of virus based on quantifying the concentration of viral genomes or viral proteins.
Historically, DNA hybridization assays were used to quantify the number of viral genomes present in a preparation. Plasmid and unpackaged vector DNA from a sample are removed with nucleases so only viral genomes inside intact capsids will be titered. The vector DNA signal is compared to the signal from the plasmid DNA standard curve and extrapolated to determine a vector genome titer.
The Optical Density (A260/280) Assay measures the concentration of viral DNA and protein. It is a physical assay measuring the concentration of viral particles (VP).
Several groups have investigated the NanoSight® device (Malvern Instruments Ltd) for viral particle quantification. This real-time nanoparticle visualization system is based on a laser-illuminated microscope technique detecting Brownian motion of virus particles in solution. While this method has utility, its technical limits are reached when host cell debris or other background particles are present.3
High-performance liquid chromatography (HPLC) is also a rapid method to quantify total viral particles since the column allows for the separation of intact virus particles from other cellular contaminants or virus particle fragments.4
The most widely used methods to quantify packaged AAV vector genomes are real-time PCR-based assays (i.e. qPCR, digital drop PCR) because they are robust, easy, fast, and convenient. The vector genome titer is determined by using appropriate primers and fluorescent probes. However, there can be substantial variability in the assay because PCR is affected by many experimental factors. This can affect the accuracy of the titer, which in turn affects the vector dosing in both preclinical and clinical settings.5 Therefore, assay design and incorporation of appropriate standards needs to be well thought out.
Infectious or Functional Titer
Infectious or functional titer is always more accurate than physical titer because it measures how much virus can infect the target cell. It is always lower than physical titer, typically by a factor of 10 to 100-fold.
However, functional titer usually takes much longer to determine and is sometimes not practical during intermediate productions stages of the viral vector production.
Functional titer is expressed as transducing units per mL (TU/mL) for lentiviral or retroviral vectors. Adenoviral vector titer is generally expressed as plaque-forming units per mL (PFU/mL) or infectious units per mL (IFU/mL). The latter term is used for viral vectors that do not lyse cell membranes and therefore are not compatible with the plaque assay.
The viral plaque assay is one of the most widely used methods to determine infectious titer. Serial dilutions of the virus are used to transduce a permissive cell line. The cells are infected at very low multiplicity of infection (MOI) to ensure that most transduced cells are infected by a single viral particle for accurate quantitation. Viruses propagate in the infected cells eventually causing cell lysis, spreading to neighboring cells eventually forming visible holes (or plaques) on the cell monolayer. Enumeration of the total plaques at particular dilution provides the number of plaque-forming units (PFU) per mL.
An Endpoint Dilution Assay (TCID50) measures the infectious virus titer by quantifying the amount of virus required to kill or to produce a cytopathic effect in 50% host cells. Although both the plaque-forming Assay and Endpoint Dilution assay give the infectious titer, they are scored by the human eye and subject to human and procedural variations.
The Immunofluorescence Foci Assay (IFA) also known as fluorescent foci assay FFA, shows good correlation with the plaque assay and captures viruses that do not lyse cell membranes (which are not compatible with plaque assays) or do not exhibit detectable cytopathic effects (to perform TCID50). IFA utilizes antibody-based staining methods to detect virally infected cells. The results of IFA are expressed in focus forming units (FFU) per ml, or IFU/mL. Overall, IFA is considered a more rapid and sensitive method to determine infectious titer than traditional plaque assays or TCID50.6
Vectors carrying reporter genes such as the green fluorescent protein (GFP) can be easily titrated by flow cytometry in transduction units (TU/mL).5
Table 1. Overview of Analytical Methods used in Viral Vector Manufacturing
| Quality | Attribute | Technique |
|---|---|---|
| Identity | Confirm presence and identity of viral vector | SDS-PAGE, Mass spectrometry (MS), Western blot (immunoblot), Genome sequencing (NGS), PCR |
| Potency | Physical viral titer | DNA hybridization, Real-time PCR (qPCR, ddPCR), Optical density (A260/280), NanoSight, HPLC |
| Functional viral titer | Plaque-forming assay, Fluorescence foci assay, TCID50 (end point dilution assay) | |
| Purity | Process-related impurities | MS, Chromatography, TEM |
| Host cell-related Impurities | Host cell DNA/RNA: Picogreen, DNA Threshold assay, qPCR, Host cell proteins: ELISA,TEM |
|
| Capsid content (empty: full capsids) | TEM, AUC | |
| Safety | Sterility | Standard sterility tests (EP 2.6.1, USP71) |
| Endotoxin | LAL method (EP 2.6.14, USP85), Rabbit pyrogen assay | |
| Mycoplasma | PCR, Cell cultured based-assays | |
| Replication Competent Virus (presence of rep or cap sequences) | Southern blotting, qPCR | |
| Adventitious Agents | In vivo and in vitro assays | |
| Stability | pH | Potentiometry |
| Osmolality | Osmometry | |
| Aggregate formation | Light microscopy, DLS, SEC-MALS, TEM, AUC, FFF-MALS |
Purity
Impurities can be derived from the host cell system within which the vector product is generated or from the downstream vector purification. Sources of host cell-related impurities are residual host cell proteins and nucleic acids derived from the production cells while other are process-related residuals from the cell culture medium (i.e. bovine serum albumin) and downstream purification processes (i.e. detergents and chromatography resin components). It is important to establish a good understanding of potential residuals early on in process development to establish acceptable tolerances in the final product.
Host Cell-Related Impurities
The quantification and removal of host-cell impurities is important since certain host cell molecules can have toxic effects in the final drug product or can act as an adjuvant to stimulate an anti-vector immune response.
Host cell nucleic acids (DNA/RNA) can be copurified with the viral vector product during production. PicoGreen and DNA Threshold Assays are two of the historical methods to determine residual DNA levels. More recently, quantifying host cell nucleic acids has been done using real-time or quantitative PCR (qPCR).
Detection of host cell-associated proteins is typically done with ELISA, where antibodies react with host proteins. One important point to consider is that certain host proteins are not recognized by commercial ELISA kits. For example, there are many variants of HEK293 cells used for virus production that can potentially express proteins not detectable by standard kits. Host cell protein impurities can vary in quantity and proportion from lot to lot during production.
These impurities can be also be successfully detected using transmission electron microscopy (TEM). MiniTEM™ by Vironova, is a novel, benchtop TEM platform that can inform on quality attributes during process development and be easily incorporated into existing QC workflows. The sample is prepared, mounted to a grid support with negative staining (to improve contrast) prior to microscopic analysis to detect impurities (i.e. proteasomes and host cell debris). The advantage over traditional TEM is that image acquisition and, more importantly image analysis, from the MiniTEM™ is automated allowing for the quantitative analysis of a greater total number of particles for more accurate and statistically significant results in shorter time.
Process Impurities
Process impurities are usually present in trace amounts but it is important that they meet pre-set safety guidelines. Mass Spectrometry (MS) and chromatography methods are widely used to identify detergents and organic solvents in the vector preparation.
The quality of vector preparation can also be analyzed through direct visualization with MiniTEM by Vironova. The system can assess viral particle morphology such as capsid integrity and detect particle aggregation.
Capsid Content
During the production of viral vectors, there exists a population of capsids that have failed to package the vector DNA. These empty capsids can represent up to 90% of the crude harvest for rAAV vector preparations.5,7 In addition, incomplete encapsidation is also observed, leading to capsids containing truncated vector genomes or capsids that contain illegitimate, non-vector DNA (from plasmids, cells, or helper viruses). While their impact on the efficacy of gene therapy is not fully understood, these capsids are considered an undesirable quality attribute5. They are a source of unnecessary, potentially antigenic material, which could elicit an unwanted immune response in the patient. For example, in a clinical study for hemophilia B, capsid-specific cytotoxic T lymphocytes were implicated in an adverse immune response after successful expression of human coagulation factor IX using recombinant AAV2-mediated gene transfer.7 Therefore, the capsid content (ratio of empty to full capsids) is an important attribute to monitor in viral vector preparations. There are two main methods under investigation to evaluate capsid content: transmission electron microscopy (TEM) and analytical ultracentrifugation (AUC).
TEM (cryoTEM): Cryogenic TEM is a method where the biological specimens are vitrified by rapid freezing in liquid ethane thereby preserving their native structure. Analysis by cryoTEM combined with Vironova Analyzer Software (VAS), a stand-alone 21 CFR part 11 compliant software for TEM based image analysis of nanoparticles, is a GMP-validated method to identify and relatively quantify empty and fully packed particles. When imaged, there is a clear, morphological distinction between packed and empty viral particles where the empty particles appear as open circles with well-defined edges and no internal density and packed particles appear circular with a dense inner core. Currently, CryoTEM™ is performed as a service by Vironova.
AUC: Analytical ultracentrifugation (AUC) is a powerful tool to distinguish and quantify different AAV species by either mass (sedimentation velocity) or density (sedimentation equilibrium). Beckman Coulter introduced the first commercial AUC sample characterization instrument. Their newest platform, the Optima AUC is the most sensitive and robust to date, offering multi-wavelength absorbance capabilities to properly quantify both genomic DNA and viral capsid content in a single experiment.
AUC is a technique that monitors the sedimentation of particles over time under a centrifugal field, providing critical information on particle molecular weight, homogeneity, and interactions with other particles and itself. The empty capsids have a different density and/or mass than the correctly packaged, full particles and those partially filled, incomplete particles allowing baseline separation on the basis of hydrodynamics under centrifugal force. The main advantages are that it is highly reproducible, can quantify viral particles in the formulation buffer (no sample preparation required), works independent of the serotype or the size and type of transgene (single or double stranded DNA) and also requires no standard for comparison making it easy to incorporated into the manufacturing workflow with little to no development time.

Each technique differs in the physical measuring principle and has its own strengths and weaknesses, but it is valuable to obtain complementary data from orthogonal methods to increase confidence in the data particularly for a quality attribute where regulatory requirements have yet to be established.
Safety
Safety testing is conducted to ensure that process intermediates or final product is free of detectable contaminating agents that can pose risks to patients. The key parameters are sterility, endotoxin, mycoplasma, adventitious virus and the presence of replication-competent virus.
Sterility testing to assess bacterial or fungal burden of the product should be performed according to USP or EP recommendations.5,8
Endotoxin can be detected using the Limulus Amebocyte Lysate (LAL) Assay or the rabbit pyrogen test. The presence of mycoplasma DNA can be detected by PCR-based methods in Vero cells or cell culture-based methods where the viral vector preparation is inoculated into growth medium to detect any growth of mycoplasma.
Mycoplasma testing should be performed on the product at the manufacturing stage when the test is most likely to detect contamination; such as after pooling of cultures for harvest, but prior to cell washing to ensure that it is not present in the preparation prior to downstream purification steps that could filter it out.
The in vivo and in vitro assays to test for adventitious virus are designed to detect the presence of infectious viral agents of human or animal origin. The in vitro assay can be performed using a number of cell lines (e.g. human, murine, bovine, porcine, etc) while rodent and embryonated hen’s eggs are commonly used for the in vivo assay.9
While viral vectors are engineered to be replication defective, generation of replication-competent virus can still occur during vector manufacturing by means of spontaneous recombination events within the producer cells. This is obviously undesirable because many viral vectors are created from pathogenic viruses posing a significant safety concern for gene therapy. Therefore, it is important to test for the presence of potential replication-competent virus in the vector preparation prior to final product release.
For quantification of replication competent AAV (rcAAV), the vector sample is incubated on a permissive cell line in the presence of adenovirus or an alternative source for helper factors.10 After three rounds of infection using cell lysate from the previous round, amplification of AAV rep or cap sequences is detectable by Southern blotting or qPCR. Vector production systems currently in use have been optimized to reduce rcAAV to levels ≤1 rcAAV per 108 vector genomes.11
Stability Testing
Stability testing of the purified viral vector at the proper storage temperature, formulation, and fill volume, and in the container used for patient doses (i.e. final product) should be performed to ensure that quality attributes are maintained and that the quantitative values (i.e. infectivity titer) are not adversely affected over the span of its shelf-life. This can include examining physical parameters such pH/ osmolality or the presence of protein aggregates. If these unwanted aggregates are not removed, they will become part of the finished biopharmaceutical drug product and may alter its bioactivity and/or cause adverse biological effects when administered to a patient (i.e. immunogenicity).
Aggregates can vary greatly in size from a few nm (subvisible) to a few mm (visible) in diameter and their formation during production, storage, shipment can be caused by numerous stressors.11 Since no single technique is able to cover this large size range, a combination of several techniques is typically necessary to evaluate this parameter. These techniques rely on different separation and detection principles ranging from basic techniques such as visual or microscopic inspection (visible particles), to more sophisticated methods to detect subvisible particles, such as dynamic light scattering (DLS), size exclusion chromatography (SEC), TEM, AUC and field flow fractionation with multi-angle static light scattering (FFF-MALS).
SEC is a long-standing industry standard as an inexpensive, high throughput method to quantify aggregates at various steps of protein product development. Ideally, SEC separates proteins and their aggregates based on their hydrodynamic volume. However, numerous factors have impacted the accuracy of analysis using SEC that typically arise from nonspecific interactions between the aggregates and the chromatographic material in the HPLC column or the column frits.11
More recently, hydrodynamic techniques such as FFF-MALS and AUC are being implemented as orthogonal methods to validate SEC aggregation data. SV (sedimentation velocity)-AUC is a robust tool to quantify aggregates because, unlike SEC, matrices are not involved in the analysis and the aggregate content often can be analyzed in the original protein formulation.11 In addition to supporting efforts in the development of accurate SEC methods, SV-AUC can be used to generate extended biophysical characterization information on potential biopharmaceuticals to support formulation/process development and comparability studies.
Conclusion
This article has outlined an extensive list of analytical tools that can be applied to different stages of viral vector manufacturing. However, it should be emphasized here that not all of the analytical controls are suitable for every stage of product development and the extent/amount of testing should reflect the stage of development. A distinction can be made between techniques that would be routinely used in a QC testing for product release versus those for extended characterization during product development, whether it be due to the reproducibility or expertise/time differences required to execute the assays. For example, applying extended characterization assays during formulation and process development is necessary to gain product knowledge and to establish specifications early on, but often times, there is a limited quantity of the final, purified viral vector which may influence which assay and how many are needed to provide sufficient data on key quality parameters for an acceptable production lot.
As the cell and gene therapy field focuses on product commercialization, it is prudent for manufacturers and therapy owners to ensure all processes and procedures are adapted to meet the current regulatory requirements. This poses challenges since the parameters for what makes a cell and gene therapy product safe for patients are still evolving and the needs of the field are diverse. It is also vitally important to harmonize analytical methods to ensure quality attributes meet acceptance criteria for a specific application (this can vary based on the disease and method of delivery into the patient). Some of the analytical technologies described may still need fine-tuning but show that there is a vested interested to ensure the safest product possible can be manufactured for these therapies. Thus, improving viral vector production must be regarded as a collaborative effort of stakeholders and regulatory agencies.
This article is the fifth in a series that was published in our eBook, “Key Considerations for Gene Therapy Manufacturing for Commercialization“. You can download all the articles in the series, by downloading the eBook below.
Footnotes
-
1. Kramberger, P., L. Urbas, and A. Strancar, Downstream processing and chromatography based analytical methods for production of vaccines, gene therapy vectors, and bacteriophages. Hum Vaccin Immunother, 2015. 11(4): p. 1010-21.
-
2. D’Costa, S., et al., Practical utilization of recombinant AAV vector reference standards: focus on vector genomes titration by free ITR qPCR. Mol Ther Methods Clin Dev, 2016. 5: p. 16019..
-
3. Heider, S. and C. Metzner, Quantitative real-time single particle analysis of virions. Virology, 2014. 462-463: p. 199-206.
-
4. Kuhn, I., et al., High-performance liquid chromatography method for rapid assessment of viral particle number in crude adenoviral lysates of mixed serotype. Gene Ther, 2007. 14(2): p. 180-4.
-
5. Dorange, F. and C. Le Bec, Latest developments in vral & non-viral vector manufacturing. Cell GeneTherapy Insights, 2018. 4(2): p. 119-129.
-
6. Kumar, P., Methods for rapid virus identification and quantification. MATER METHODS, 2013. 3: p. 207.
-
7. Wright, J.F., Transient transfection methods for clinical adeno-associated viral vector production. Hum Gene Ther, 2009. 20(7): p. 698-706.
-
8. Husain, S.R., et al., Gene therapy for cancer: regulatory considerations for approval. Cancer Gene Ther, 2015. 22(12): p. 554-63.
-
9. Mandel, R.J., C. Burger, and R.O. Snyder, Viral vectors for in vivo gene transfer in Parkinson’s disease: properties and clinical grade production. Exp Neurol, 2008. 209(1): p. 58-71.
-
10. Schnodt, M. and H. Buning, Improving the quality of adeno-associated viral vector preparations: the challenge of product-related impurities. Hum Gene Ther Methods, 2017. 28(3): p. 101-108.
-
11. den Engelsman, J., et al., Strategies for the assessment of protein aggregates in pharmaceutical biotech product development. Pharm Res, 2011. 28(4): p. 920-33